
氯离子清除试验在Gitelman综合征鉴别诊断中的应用
2016-08-02 06:39:51彭晓艳蒋兰萍李乃适童安莉邢小平李雪梅李学旺陈丽萌
中国医学科学院学报 2016年3期
彭晓艳,蒋兰萍,袁 涛,乐 偲,郑 可,王 鸥,李乃适,李 伟,童安莉,邢小平,李雪梅,李学旺,陈丽萌
中国医学科学院 北京协和医学院 北京协和医院 1肾内科 2内分泌科国家卫生和计划生育委员会重点实验室,北京 100730
·论著·
氯离子清除试验在Gitelman综合征鉴别诊断中的应用
彭晓艳1,蒋兰萍1,袁涛2,乐偲1,郑可1,王鸥2,李乃适2,李伟2,童安莉2,邢小平2,李雪梅1,李学旺1,陈丽萌1
中国医学科学院北京协和医学院北京协和医院1肾内科2内分泌科国家卫生和计划生育委员会重点实验室,北京 100730
摘要:目的评估氯离子清除试验在Gitelman综合征(GS)鉴别诊断中的应用价值。方法以临床低钾、碱中毒、疑诊为GS的患者为研究对象,提取外周血DNA进行SLC12A3基因筛查作为金标准。按照标准操作流程进行氢氯噻嗪试验和速尿试验,测定患者基线和用药后3 h内氯离子排泄分数改变量的最大值(ΔFECl),评价对氢氯噻嗪和速尿的反应性,并与健康受试者比较,绘制受试者工作特征(ROC)曲线确定ΔFECl绝对值和相对值诊断GS的截点,计算氯离子清除试验诊断GS的灵敏度和特异度。结果27例患者和20例健康受试者进行了氢氯噻嗪试验。以SLC12A3基因检测为诊断金标准,23例患者被诊断为GS。以ΔFECl绝对值和相对值诊断GS的ROC曲线下面积分别为0.987(95%CI:0.963~1.000,P<0.001)和0.984(95%CI:0.950~1.000,P<0.001)。选择适当的截点,灵敏度和特异度均达到95%以上。8例患者同时完成了氢氯噻嗪试验和速尿试验,其中5例患者对氢氯噻嗪无反应(ΔFECl绝对值≤2.86% 或相对值≤223%),对速尿反应敏感,同时存在SLC12A3基因突变,确诊为GS;3例患者对氢氯噻嗪反应敏感,而使用速尿后FECl改变不明显,基因测序除外GS,考虑Bartter综合征可能性大。结论临床上综合应用氢氯噻嗪试验和速尿试验观察氯离子排泄分数的变化能有效鉴别GS和BS患者。
关键词:Gitelman综合征;氯离子清除试验;诊断试验
ActaAcadMedSin,2016,38(3):275-282
Gitelman综合征(Gitelman syndrome,GS)是一组罕见的常染色体隐性遗传病,由编码远端肾小管噻嗪敏感的钠-氯共转运子(sodium-chloride cotransporter,NCC)的SLC12A3基因突变所致[1],多于青少年起病,临床主要表现为低钾、代谢性碱中毒,常伴有低血镁和低尿钙,肾素-血管紧张素-醛固酮系统活化,但血压正常或偏低[2]。与GS临床表现相似的另一类遗传性失盐性肾病是Bartter综合征(Batter syndrome,BS)[1],由编码髓袢升支粗段的钠-钾-氯共转运子2(sodium-potassium-chloride cotransporter- 2,NKCC2)及其相应的调节蛋白基因突变导致,分为5型,受累基因包括SLC12A1(编码NKCC- 2)、KCNJ1、CLCNKB、BSND及CASR[3],发病更早,病情更严重,血镁和尿钙不低[4]。作为遗传性失盐性肾病,基因诊断是金标准[5],但其检测成本高、操作复杂,难以在临床中推广;另一方面因存在基因调控区域变异、转录修饰、外显率等问题,现有外显子测序的方法难以确诊所有GS和BS患者[6]。应用直接抑制突变基因编码NCC和NKCC2蛋白功能的药物—氢氯噻嗪(hydrochlorothiazide,HCT)和速尿(furosemide,FUR),可以直接评价靶蛋白的生理功能[7],临床诊断GS和BS[8]。目前国内采用氯离子清除试验诊断GS的病例较少[9- 10],且缺少正常对照[11]。本研究采用简化的氯离子清除试验,与基因诊断的金标准比较,验证其用于临床诊断GS的效率。
对象和方法
对象2011年11月至2015年7月以原因不明“低钾血症”在北京协和医院就诊的临床可疑GS患者,临床符合以下特点:(1)能够根据病史,除外饮食钾摄入不足、消化道失钾或利尿剂继发的低钾,除外钾分布异常;(2)存在肾性失钾:即血钾小于3.0 mmol/L时,24 h尿钾大于20 mmol;或血钾小于3.5 mmol/L时,24 h尿钾大于25 mmol;(3)血压正常或偏低(<140/90 mmHg)(1 mmHg=0.133 kPa);(4)代谢性碱中毒[12]。以同期招募的性别与年龄匹配的健康志愿者为对照。本研究经北京协和医院伦理委员会批准,所有受试者和健康志愿者均签署知情同意书。
数据采集清晨抽取空腹血测定血肌酐、电解质等生化指标;同期留取24 h尿标本测定尿肌酐和电解质。采用放射免疫方法检测立位血浆肾素活性(plasma renin activity,PRA)、血管紧张素Ⅱ(angiotensin Ⅱ,AngⅡ)和醛固酮(aldosterone,Ald)水平。常规采集心电图。抽取患者外周EDTA抗凝血,采用QIAGEN试剂盒按照说明书操作,提取血细胞DNA,普通PCR扩增SLC12A3基因的26个外显子,于华大基因公司进行直接测序法筛查基因突变,并与NCBI数据库比对,作为诊断的金标准[13]。
氯离子清除试验所有患者在进行氯离子清除试验前均停用利尿剂1周以上,但补钾、补镁的药物仅需在试验前1 d停用即可,血钾纠正至3.0 mmol/L以上方可进行试验。HCT试验与FUR试验最好间隔1周进行。饮食不限制。
参考文献[7]及本实验室前期确立的方法进行氢氯噻嗪试验和速尿实验[14],具体为:患者空腹一晚后,15 min内饮完普通饮用水(10 ml/kg体重)以促进自发性排尿,以后每1 h饮水150 ml。在第30和60分钟采集尿液,第60分钟采血,计算基线平均电解质排泄分数,并口服HCT 50 mg(HCT试验,儿童1 mg/kg体重,不超过50 mg)或肌注FUR 20 mg(FUR试验)。以后每30 min采集1次尿液,共6次;最后一次采集尿液时采血。试验中除排尿时可站立外,要求保持自然卧位4 h。
溶质X的排泄分数 (FEX)=100×(UX/SX)×(Scr/Ucr),Scr=血肌酐浓度,Ucr=尿肌酐浓度,X指Na、K、Cl。取第2次血和后6次尿液计算服药后的排泄分数,取最大值减去基础排泄分数,即为服药后排泄分数的差值(ΔFEX),计算相对基线水平增加的比例。
统计学处理采用SPSS 20.0统计软件,计量资料采用均数士标准差表示,组间比较采用独立样本t检验,非正态分布时采用非参数检验。纳入所有接受过HCT试验的患者和健康受试者,绘制ROC曲线,计算曲线下面积,确定ΔFECl绝对值或相对值诊断GS的最佳截点,计算灵敏度、特异度和符合率,Kappa值评价HCT试验与金标准的一致性。P<0.05为差异有统计学意义。
结果
患者纳入及基因测序结果本研究共27例患者完成了HCT试验,其中,23例经基因筛查诊断为GS,4例未发现突变。8例患者同时完成了HCT试验和FUR试验,SLC12A3基因检测提示其中5例存在基因突变:4例为复合杂合突变(3例携带2种突变,1例携带3种突变),1例纯合突变。此8例患者常见的临床表现依次为乏力(87.5%),夜尿(75%),手足痉挛、抽搐(62.5%),心悸(62.5%),肌肉疼痛(50%)等,1例患者有过一过性意识丧失、跌倒(表1)。8例患者血压均正常,都曾表现为低钾血症,经补钾治疗后,6例患者仍存在低钾血症,1例患者低血镁。所有患者血钠、血钙和血肌酐基本正常。7例患者尿钾排泄显著升高,6例尿钙肌酐比值降低。67.5%的患者存在明显碱中毒,表现为动脉血气pH>7.45,碳酸氢根离子浓度大于26 mmol/L。5例患者存在不同程度的肾素、血管紧张素Ⅱ和醛固酮水平增加(表2)。
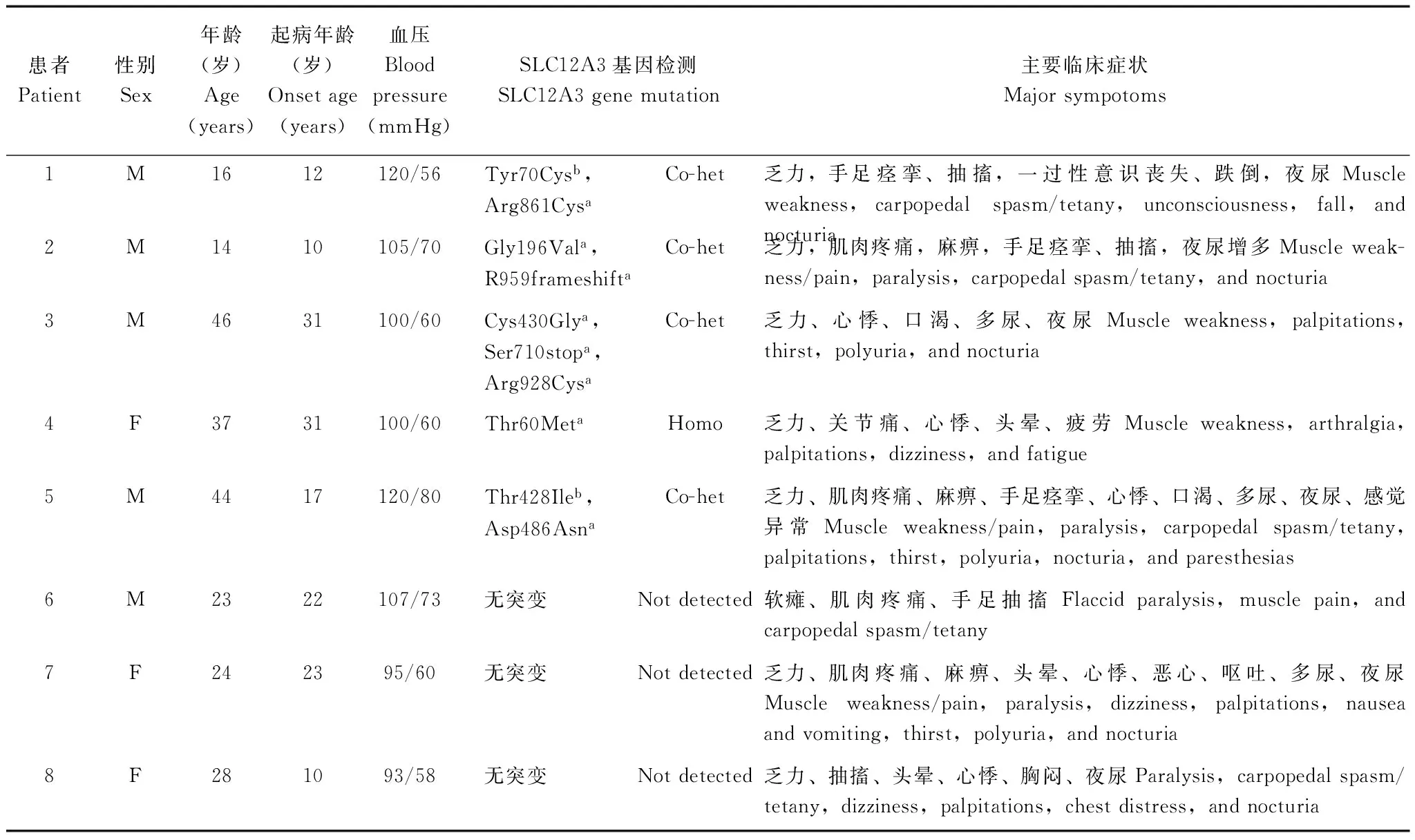
表 1 8例患者一般情况及SLC12A3基因检测结果
M:男;F:女;Homo:纯合突变;Het:杂合突变;Co-het:复合杂合突变;a:突变为文献已报道;b:突变为本研究新发现,经PolyPhen- 2、mutationtaster等预测该突变可能致病;1 mmHg=0.133 kPa
M:male;F:female;Homo:homozygosity;Het:heterozygosity;Co-het:compound heterozygosity;a:these mutations had been reported in previous studies;b:new mutations detected in this study,as predicted to be probably pathogenic by PolyPhen- 2,mutationtaster;1 mmHg=0.133 kPa

表 2 8例患者临床资料
ABE:实际碱剩余;PRA:血浆肾素活性;RAAS:肾素-血管紧张素-醛固酮系统;AngⅡ:血管紧张素Ⅱ;Ald:醛固酮;PTH:甲状旁腺素;血/尿电解质同期检测,正常参考值范围来自正常人群
ABE:actual base excess;PRA:plasma renin activity;RAAS:renin-angiotensin-aldosterone system;AngⅡ:angiotensin Ⅱ;Ald:aldosterone;PTH:parathyroid hormone;the blood and urine were tested nearly the same time,reference values for the biochemical parameters were based on the general population
HCT试验结果5例GS患者的氯离子排泄分数基础值明显高于对照组[(1.63±0.69)% 比 (0.88±0.34)%,P=0.002];口服HCT后,增加量明显低于对照组[(1.44±1.67)%比(4.46±1.04)%,P<0.001]。3例疑诊BS患者使用HCT后,钠、氯、钾离子排泄分数均明显增加(图1),其中1例患者用药后ΔFENa和ΔFECl分别达到9.30%和14.11%,高于正常人对HCT的反应水平(表3)。
FUR试验结果GS患者使用FUR后ΔFECl均超过20%。其中,患者1为82.38%,是正常人的4倍,相对自身基线水平增加了59倍;其钠离子和钾离子的改变量也高于健康对照(表4)。BS患者使用FUR后FECl的最大值低于11%,ΔFECl分别为10.61%、1.9%和6.87%,反应弱于正常人(表4,图1)。
氯离子清除试验的敏感性和特异性以基因诊断结果作为金标准,ΔFECl的绝对值和相对值用于诊断GS的ROC曲线下面积分别为0.987(95%CI:0.963~1.000,P<0.001)和0.984(95%CI:0.950~1.000,P<0.001)。当ΔFECl绝对值截点选择2.86%时,HCT试验诊断GS的灵敏度为95.7%,特异度为95.8%,符合率为95.7%,与基因诊断具有较好的一致性(Kappa值=0.915,P<0.001)。选择ΔFECl相对值223%作为截点时,HCT试验诊断GS的灵敏度为95.7%,特异度为100%,符合率为97.9%,与基因诊断具有较好的一致性(kappa值=0.957,P<0.001)。
8例患者中,3例ΔFECl绝对值小于2.86%,相对值小于223%,患者对HCT反应减弱,临床诊断为GS,基因检测确诊;3例根据ΔFECl绝对值和相对值判断远曲小管NCC功能正常,在FUR试验中反应弱于正常人,符合BS;2例患者在HCT试验中ΔFECl绝对值和相对值各有1项低于正常值,FUR试验中均反应良好,经基因检测确诊为GS。
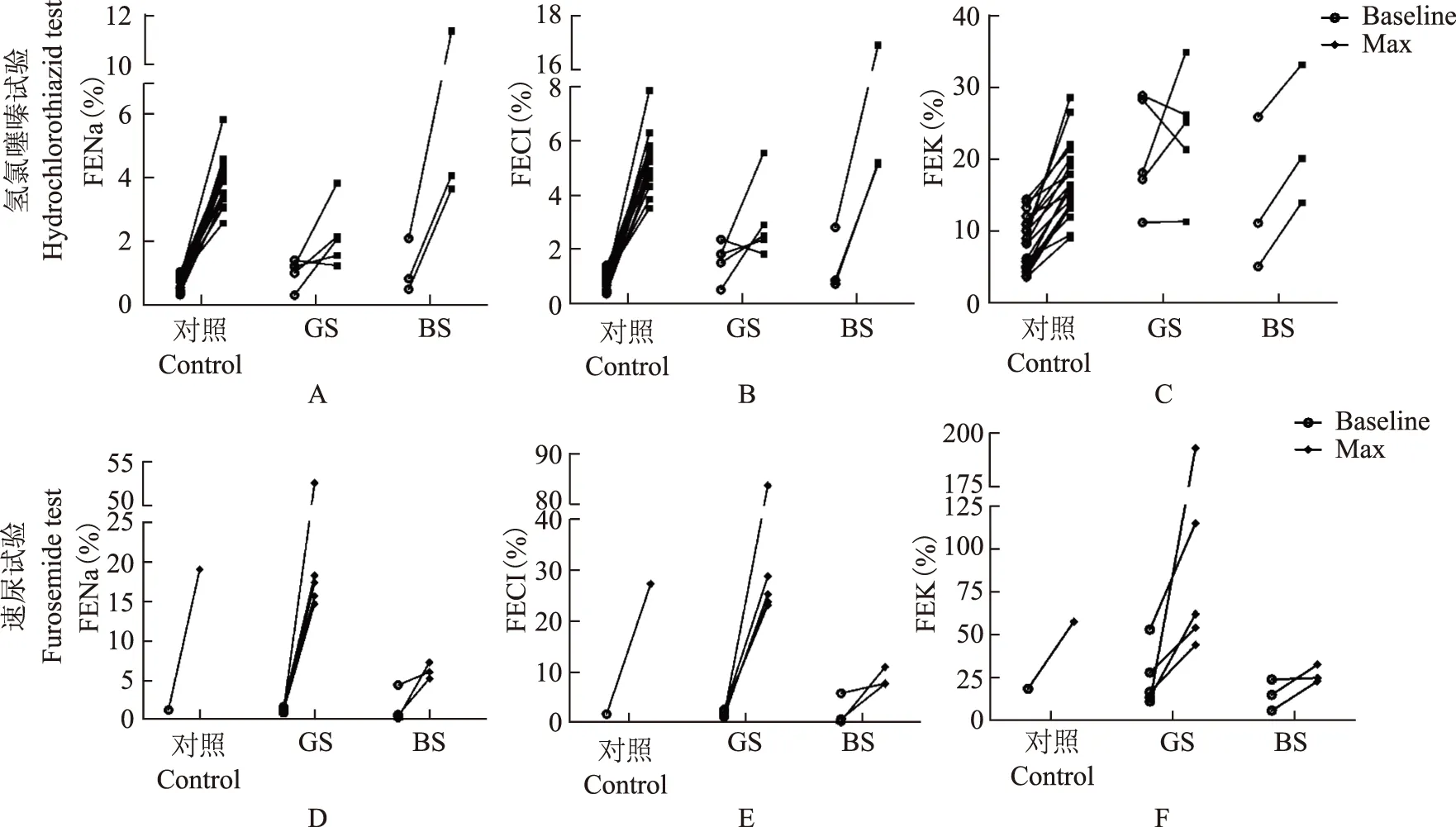
FENa:钠排泄分数;FECl:氯排泄分数;FEK:钾排泄分数;GS:Gitelman综合征;BS:Batter综合征;氢氯噻嗪试验的对照值来自参考文献[14],速尿试验的对照值来自参考文献[7];GS组含5例基因确诊的GS患者,BS 组含3例无SLC12A3基因突变被临床诊断为BS的患者
FENa:sodium excretion fraction;FECl:chloride excretion fraction;FEK:potasium excretion fraction;GS:Gitelman syndrome;BS:Bartter syndrome;for hydrochlorothiazid test,the data of healthy controls were from reference [14];for furosemide test,the data of controls group were from reference [7];genetically confirmed GS patients were described as GS group (n=5),three patients without SLC12A3 gene mutation and clinically diagnosed as BS were in BS group
A~C.氢氯噻嗪试验结果;D~F.速尿试验结果
A-C.hydrochlorothiazide test;D-F.furosemide test
图 1氢氯噻嗪试验和速尿试验结果
Fig 1The individual hydrochlorothiazide test and furosemide test
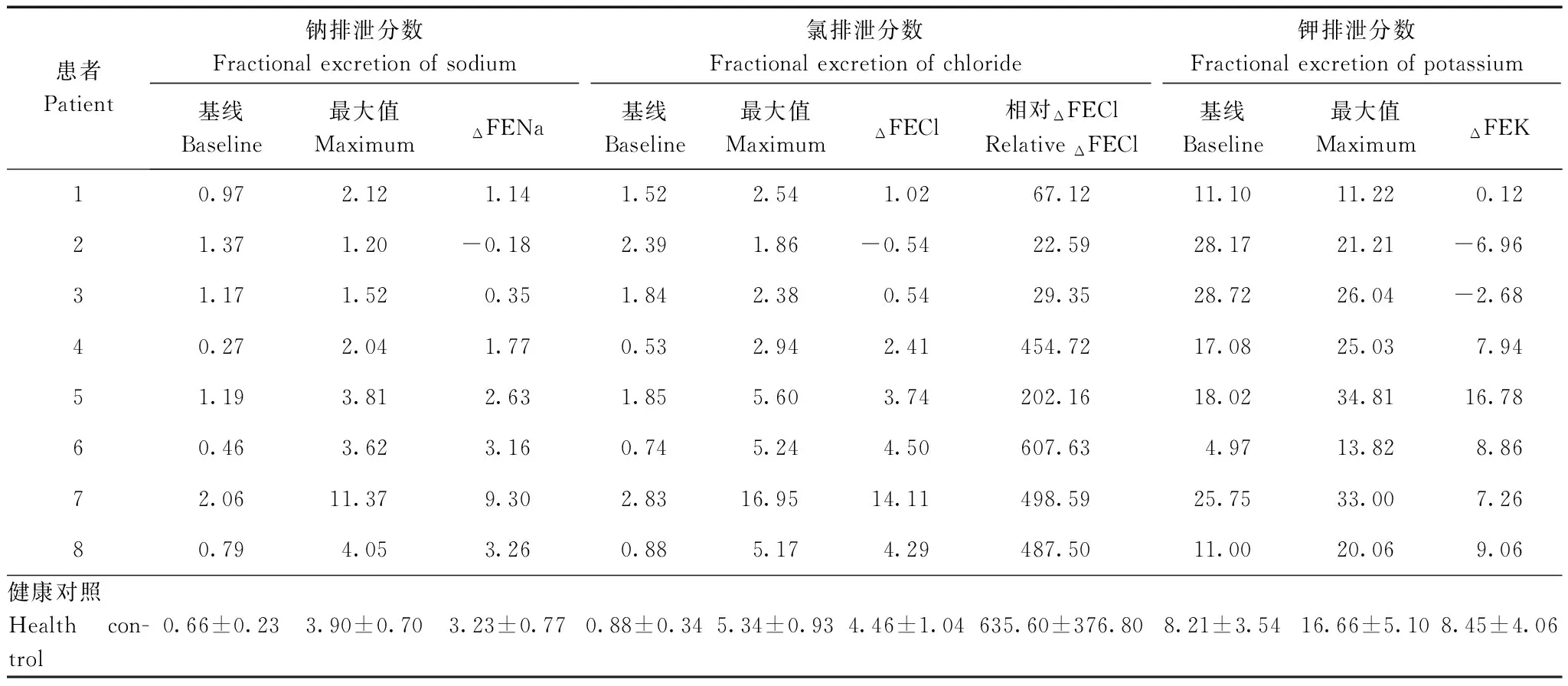
表 3 氢氯噻嗪试验结果(%)
ΔFENa:钠排泄分数的改变量;ΔFECl:氯排泄分数的改变量;ΔFEK:钾排泄分数的改变量;对照值来自参考文献[14]
ΔFENa:increasement of sodium excretion fraction;ΔFECl:increasement of chloride excretion fraction;ΔFEK:increasement of potasium excretion fraction;for HCT test,the value of healthy controls were from reference [14]

表 4 速尿试验结果(%)
对照值来自参考文献[7]
The data of controls group were from reference[7]
讨论
GS由编码远曲小管噻嗪敏感的NCC的SLC12A3基因突变所致[1],多于青少年起病,主要表现为低钾、代谢性碱中毒,肾素-血管紧张素-醛固酮系统活化,但血压正常或偏低,因远曲小管决定镁离子的最终排泄[3],因此GS常伴有低血镁[2,4]。过去认为GS患病率低、疾病本身的危害小,且治疗手段有限,目前这一观点正在转变[15- 16]。Vargas-Poussou等[17]报道了448例非亲缘关系的GS患者,检测出172种突变。王朝晖等[18]回顾性总结3年内新发现的家族性肾脏疾病家系,发现GS多达37个,仅次于家族性IgA肾病(58个)、Alport(56个)和家族性局灶性节段性肾小球硬化(45个)[19]。70%的GS患者同时存在3个以上系统(骨骼肌、肾脏、胃肠道、心血管和神经)的临床症状,慢性肾功能不全和糖尿病患病率高、发病年龄提前[16,20],GS患者发生晕厥、严重心律失常或进展为终末期肾病的病例也有报道。由此可见,提高GS的诊断率将有助于人们深入认识该疾病的病理生理机制,寻找可能的治疗方法。
GS曾被当成BS的一个亚型[21],后者主要由编码髓袢升支粗段的NKCC2及其相应的调节蛋白基因突变导致,因髓袢升支粗段对于水和盐的重吸收作用较远曲小管更关键,因此BS发病更早,临床鉴别诊断依据主要是GS存在低血镁和低尿钙[22]。基因检测的快速发展提高了GS的诊断率,但作为金标准[5],其临床推广仍然有局限性。一方面,基因检测成本高、操作复杂、难以发现内含子深处和基因转录调控区域的突变[17,23- 24]。另一方面,大部分GS患者携带的是复合杂合突变[17],难以通过针对单一突变的体外表达体系判断患者的NCC蛋白功能;携带相同突变的不同患者,临床表现各异[25];同一患者在不同阶段NCC蛋白的功能也可能存在差异。因此应用小剂量能直接抑制NCC和NKCC2蛋白功能的药物如HCT和FUR,建立临床生理功能试验能更直接鉴别GS和BS[8]。本研究在可疑GS患者中开展简化的氯离子清除试验,与基因诊断比较,评价了HCT试验联合FUR试验诊断GS的效率,以期为其临床推广提供依据。
最初主要用水负荷试验来鉴别诊断BS[26];随后用低渗盐水负荷后的利尿剂刺激试验从可疑BS中成功诊断了GS[27],但操作方法复杂[26- 31]。1997年,Colussi等[7]对11例临床诊断的GS和23例对照进行了简化的HCT和FUR试验,发现GS患者对HCT(6例)反应减弱,对FUR(11例)反应良好,但缺乏基因诊断金标准;直到2007年,该研究团队才在基因确诊的GS、BS和假性BS患者中进行了HCT试验,发现92.7%的GS患者对HCT无明显反应(ΔFECl<2.3%),BS及假性BS均反应良好[8]。国内应用氯离子清除试验鉴别诊断GS的病例较少且无正常对照[1,9- 10]。本课题组建立了简化的方法,并建立中国人群HCT试验的正常值[14],与基因诊断金标准进行比对,其诊断GS的灵敏度和特异度均高于95%(不同截点而异)。该方法如联合FUR试验,可鉴别绝大多数GS和BS患者,且试验简便易行、成本低、相对安全(GS患者对HCT不敏感,目前尚无容量不足、严重低血钾等报道)。除诊断价值之外,该功能试验还能反映患者NCC功能缺陷程度,后者可能与临床表现的严重程度密切相关[13- 14]。此外,该试验还可用于鉴别其他继发性远端肾小管NCC障碍,起到功能定位的作用。氯离子清除试验的原理类似于“激发试验”,理论上可能存在一定风险。故本研究中,首先根据临床高度怀疑的疾病类型来选择试验类型,GS首选HCT实验,BS首选FUR试验。其次要求将患者血钾水平纠正到3.0 mmol/L以上方可进行试验,试验当天清晨尽早开始,试验中患者血钾水平波动小于0.3 mmol/L,试验结束后,患者可尽早恢复补钾药物治疗,未出现不良事件。因此,氯离子清除试验是一种相对安全有效的诊断方法。
由于GS和BS中受累的蛋白在表达量和分布上各有差异,在功能上相互影响和协调,而电解质排泄分数只是小管各部位协调的结果,存在一定假阴性,特别是针对BS患者时变异较大。2010年,Nozu等[32]报道的氯离子清除试验结果中,SLC12A1突变后(BSⅠ型)对FUR无反应;KCNJ1突变(BS Ⅱ型)患者对两类利尿剂均反应良好[33];但CLCNKB突变的患者(BS Ⅲ型)对FUR反应正常,对HCT无反应,与Ohkubo等[34]研究结果相似。本研究中有2例患者在HCT试验中ΔFECl的绝对值和相对值各有1项正常,对FUR反应良好,基因确诊为GS,有待除外是否合并KCNJ1基因突变;3例临床诊断为BS的患者筛查了GS(SLC12A3)基因正常,筛查了CLCNKB和CaSR基因无致病突变,但遗憾的是未能筛查SLC12A1和BSND基因。
综上,本研究结果显示,氯离子清除试验(包括HCT试验和FUR试验)能够鉴别大多数GS和BS。氯离子清除试验相对于基因测序是一种简便、经济、有效诊断GS的方法,并能根据生理功能鉴别肾小管功能缺失部位,可以在临床上推广使用。
[1]Fremont OT,Chan JC. Understanding Bartter syndrome and Gitelman syndrome[J]. World J Pediatr, 2012,8(1):25- 30.
[2]杨国庆,赵蕾,席文琪,等. Gitelman综合征9例临床分析 [J].中华内科杂志,2006,45(8):650- 653.
[3]Seyberth HW,Schlingmann KP. Bartter-and Gitelman-like syndromes:salt-losing tubulopathies with loop or DCT defects[J]. Pediatr Nephrol, 2011,26(10):1789- 1802.
[4]Seyberth HW. An improved terminology and classification of Bartter-like syndromes[J]. Nat Clin Pract Nephrol, 2008,4(10):560- 567.
[5]Moes AD,van der Lubbe N,Zietse R,et al. The sodium chloride cotransporter SLC12A3:new roles in sodium,potassium,and blood pressure regulation[J]. Pflugers Arch,2014,466(1):107- 118.
[6]Riveira-Munoz E,Chang Q,Bindels RJ,et al. Gitelman’s syndrome:towards genotype-phenotype correlations[J]. Pediatr Nephrol, 2007,22(3):326- 332.
[7]Colussi G,Rombola G,Brunati C,et al. Abnormal reabsorption of Na+/CI-by the thiazide-inhibitable transporter of the distal convoluted tubule in Gitelman’s syndrome[J]. Am J Nephrol, 1997,17(2):103- 111.
[8]Colussi G,Bettinelli A,Tedeschi S,et al. A thiazide test for the diagnosis of renal tubular hypokalemic disorders[J]. Clin J Am Soc Nephrol, 2007,2(3):454- 460.
[9]冉兴无,王椿,代芳,等. 表现为严重低钙血症、周期性麻痹的Gitelman 氏综合征[J]. 四川大学学报:医学版,2005,36(4):583- 587.
[10]邵乐平. Gitelman综合征分子发病机制的研究及临床分析[D]. 上海:上海交通大学,2007.
[11]秦岭. 中国人Gitelman综合征SLC12A3基因突变及相关功能研究[D]. 上海:上海交通大学,2009.
[12]Nakhoul F,Nakhoul N,Dorman E,et al. Gitelman’s syndrome:a pathophysiological and clinical update[J]. Endocrine, 2012,41(1):53- 57.
[13]Jiang L,Chen C,Yuan T,et al. Clinical severity of Gitelman syndrome determined by serum magnesium[J]. Am J Nephrol, 2014,39(4):357- 366.
[14]Jiang L,Peng X,Ma J,et al. Normomagnesemic Gitelman syndrome patients exhibit a stronger reaction to thiazide than hypomagnesemic patients[J]. Endocr Pract,2015,21(9):1017- 1025.
[15]秦岭,邵乐平,任红,等. 中国人Gitelman综合征高发突变的基因型和表型特征[J]. 肾脏病与透析肾移植杂志, 2008,17(4):331- 334.
[16]Tseng MH,Yang SS,Hsu YJ,et al. Genotype,phenotype,and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation[J]. J Clin Endocrinol Metab, 2012,97(8):E1478-E1482.
[17]Vargas-Poussou R,Dahan K,Kahila D,et al. Spectrum of mutations in Gitelman syndrome[J]. J Am Soc Nephrol,2011,22(4):693- 703.
[18]王朝晖,谢静远,马骏,等. 遗传性肾脏疾病单中心3年临床调查[J]. 中国实用内科杂志, 2014,34(3):254- 257.
[19]Qin L,Shao L,Ren H,et al. Identification of five novel variants in the thiazide-sensitive NaCl co-transporter gene in Chinese patients with Gitelman syndrome[J]. Nephrology (Carlton),2009,14(1):52- 58.
[20]Matsunoshita N,Nozu K,Shono A,et al. Differential diagnosis of Bartter syndrome,Gitelman syndrome,and pseudo-Bartter/Gitelman syndrome based on clinical characteristics[J].Genet Med, 2016,18(2):180- 188.
[21]Unwin RJ,Capasso G. Bartter’s and Gitelman’s syndromes:their relationship to the actions of loop and thiazide diuretics[J]. Curr Opin Pharmacol, 2006,6(2):208- 213.
[22]Koulouridis E,Koulouridis I. Molecular pathophysiology of Bartter’s and Gitelman’s syndromes[J]. World J Pediatr,2015,11(2):113- 125.
[23]Takeuchi Y,Mishima E,Shima H,et al. Exonic mutations in the SLC12A3 gene cause exon skipping and premature termination in Gitelman syndrome[J]. J Am Soc Nephrol,2015,26(2):271- 279.
[24]Glaudemans B,Yntema HG,San-Cristobal P,et al. Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome[J]. Eur J Hum Genet, 2012,20(3):263- 270.
[25]Lin SH,Cheng NL,Hsu YJ,et al. Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/chloride cotransporter[J]. Am J Kidney Dis, 2004,43(2):304- 312.
[26]Puschett JB,Greenberg A,Mitro R,et al. Variant of Bartter’s syndrome with a distal tubular rather than loop of Henle defect[J]. Nephron, 1988,50(3):205- 211.
[27]Sutton RA,Mavichak V,Halabe A,et al. Bartter’s syndrome:evidence suggesting a distal tubular defect in a hypocalciuric variant of the syndrome[J]. Miner Electrolyte Metab, 1992,18(1):43- 51.
[28]Nakamura A,Shimizu C,Nagai S,et al. Problems in diagnosing atypical Gitelman’s syndrome presenting with normomagnesaemia[J]. Clin Endocrinol (Oxf),2010,72(2):272- 276.
[29]Bettinelli A,Vezzoli G,Colussi G,et al. Genotype-phenotype correlations in normotensive patients with primary renal tubular hypokalemic metabolic alkalosis[J]. J Nephrol,1998,11(2):61- 69.
[30]Kockerling A,Reinalter SC,Seyberth HW. Impaired respon- se to furosemide in hyperprostaglandin E syndrome:evidence for a tubular defect in the loop of Henle[J]. J Pediatr, 1996,129(4):519- 528.
[31]Uribarri J,Alveranga D,Oh MS,et al. Bartter’s syndrome due to a defect in salt reabsorption in the distal convoluted tubule[J]. Nephron, 1985,40(1):52- 56.
[32]Nozu K,Iijima K,Kanda K,et al. The pharmacological characteristics of molecular-based inherited salt-losing tubulopathies[J]. J Clin Endocrinol Metab, 2010,95(12):E511-E518.
[33]Al Shibli A,Narchi H. Bartter and Gitelman syndromes:spectrum of clinical manifestations caused by different mutations[J]. World J Methodol, 2015,5(2):55- 61.
[34]Ohkubo K,Matsuzaki T,Yuki M,et al. A novel mutation of CLCNKB in a Japanese patient of Gitelman-like phenotype with diuretic insensitivity to thiazide administration[J]. Meta Gene, 2014,2:342- 348.
基金项目:国家自然科学基金(81470937、81170674)和首都特色临床研究Supported by the National Natural Sciences Foundation of China(81470937,81170674)and the Capital Specialized Clinical Application Project;第一、二位作者对本文贡献一致 The first two authors contributed equally to this article
通信作者:陈丽萌电话:010- 69155058,电子邮件:chenlpumch@163.com
中图分类号:R692.6
文献标志码:A
文章编号:1000- 503X(2016)03- 0275- 08
DOI:10.3881/j.issn.1000- 503X.2016.03.006
Corresponding author:CHEN Li-mengTel:010- 69155058,E-mail:chenlpumch@163.com
(收稿日期:2015- 07- 15)
Value of Chloride Clearance Test in Differential Diagnosis of Gitelman Syndrome
PENG Xiao-yan1,JIANG Lan-ping1,YUAN Tao2,YUE Cai1,ZHENG Ke1,WANG Ou2,LI Nai-shi2,LI Wei2,TONG An-li2,XING Xiao-ping2,LI Xue-mei1,LI Xue-wang1,CHEN Li-meng1
1Department of Nephrology,2State Key Laboratory of Endocrinology,Ministry of Health,Department of Endocrinology,PUMC Hospital,CAMS and PUMC,Beijing 100730,China
ABSTRACT:ObjectiveTo investigate the value of chloride clearance test in differential diagnosis of Gitelman syndrome (GS). MethodsFor patients with hypokalemic metabolic alkalosis and highly suspected GS,clinical data were documented and SLC12A3 gene screening was performed as gold standard to diagnose GS. Hydrochlorothiazide (HCT) test and furosemide (FUR) test were performed according to the standard process. Baseline and maximal increasement of chloride excretion fraction (FECl,the net and relative increase measured asΔFECl) were compared between patients and controls to evaluated the reaction to the corresponding diuretics. Receiver operating characteristic (ROC) curve was used to evaluate the sensitivity and specificity of HCT test in GS diagnosis. ResultsTotally 27 patients and 20 health controls received HCT test. Among those patients,23 were diagnosed with GS genetically. When using the net and relativeΔFECl to diagnose GS,the areas under the ROC curve were 0.987 (95% CI∶0.963~1.000,P<0.001) and 0.984 (95%CI∶0.950~1.000,P<0.001),respectively. When a reasonable cutoff value forΔFECl was selected,the sensitivity and specificity were both higher than 95%. Eight patients received both HCT test and FUR test. Five of them showed decreased reaction to HCT(netΔFECl≤2.86% or relativeΔFECl≤223%),while normal reaction to FUR.SLC12A3 mutations confirmed their GS. Three patients with blunt reaction to FUR showed normal reaction to HCT,finally they were diagnosed as BS clinically because no SLC12A3 gene mutation was detected. ConclusionComprehensive application of HCT test and FUR test to evaluate the diuretic reaction can effectively differentiate GS and BS.
Key words:Gitelman syndrome;chloride clearance study;diagnostic test
