
结核分枝杆菌分子分型技术的研究和应用
2016-07-28 01:57:45李道群夏雪山宋玉竹张阿梅
中国人兽共患病学报 2016年1期
关键词:结核分枝杆菌
李道群,夏雪山,宋玉竹,张阿梅
结核分枝杆菌分子分型技术的研究和应用
李道群1,2,夏雪山1,2,宋玉竹1,2,张阿梅1,2
1.昆明理工大学生命科学与技术学院,昆明650500;2.云南省分子医学研究中心,昆明650500
摘要:结核分枝杆菌是引发结核病的病原菌。近年,结核病发病有增加趋势,特别是结核杆菌耐药株增多,这些都提示其分子流行病学特点发生了改变。对不同基因型的结核病患者选取不同的治疗方案,是当今个性化医疗和结核病有效治疗的必然要求。目前广泛使用的结核分枝杆菌基因分型方法主要有插入序列6110限制性片段长度多态性、间隔区寡核苷酸分型法、多位点串联重复序列分析、全基因组测序技术、耐药相关基因突变谱分析、基因芯片分型技术等,这些方法在临床和基础研究中的应用各有利弊。本文对结核分枝杆菌的基因分型技术和应用作一综述,以期为临床工作者和基础研究人员选择适合的分型技术提供理论依据。
关键词:结核分枝杆菌;分型技术;基因分型
Supported by the Yunnan Science and Technology Commission(Nos. 2012AE003 and 2013BC009) and the Training Program Foundation for Talent of Kunming University of Science and Technology(No. KKSY201226148)
目前,结核病仍然是危害全人类健康的重要感染性疾病之一[1]。近年来由于流动人口增加、结核耐药菌株增多、HIV共感染的加剧,使得结核病的发病率和死亡率呈现逐年攀升的趋势。2013年全球结核死亡患者达到130万,亚洲地区就达占到58%(仅我国就占到全球的10%左右)。而传统的流行病学研究已不能很好地揭示其传播规律,因此分子生物学与流行病学研究方法相结合产生的分子流行病学方法在结核分枝杆菌分型方面表现出明显优势。基因分型就是利用这些基因序列多样性对分枝杆菌菌株间差异进行区分,以了解结核病传播、病原演变情况、寻找结核患者耐药产生原因、分析流行病学事件(如:社会性暴发和医源性感染),并且对于结核病的预防、治疗和控制具有重要的意义。提供简便、快速、特异、适合临床应用的检查技术是目前研究的重点之一,所以本文就国内外建立的的MTB基因分型技术和应用进行综述。
1基于重复序列IS6110的限制性片段长度多态性分型技术
结核分枝杆菌基因组中存在许多DNA重复序列,常用于结核分型研究的重复片段主要为IS6110和IS1081[2-8](表1)。IS6110是一段自然发生的转座序列,能够进行自我复制,属于IS3家族成员,通常为8~20个拷贝,但也存在零拷贝数的情况[5]。IS1081出现在于牛结核分枝杆菌中,属于IS256家族,通常有5~7个拷贝,“指纹”图谱具有多态性[6]。Collins等[9]在牛结核分枝杆菌中发现了IS1081片段,该片段与IS61110不同,在人型复合分枝杆菌中拷贝数仅为6个。IS1081在牛结核分枝杆菌中具有多态性,因此可以作为区分牛型与人型结核分枝杆菌的鉴定依据。IS6110序列拷贝数之间的差异构成了结核分枝杆菌特有的“指纹”图谱。该分型方法是先提取结核杆菌DNA,用限制性内切酶PvuII进行消化;经过电泳分离(可产生16-40个条带)后,将片段转移到杂交膜上,用标记的探针与之杂交。与探针杂合的膜上的DNA片段即可被检测到,最终形成RFLP“指纹”图谱[7-8,10]。虽然IS6110-RFLP(Isertion-element 6110 restriction fragment length polymorphism)是经典的结核分枝杆菌基因分型技术,但是有自身的局限性,如样本要求量大(≥3 μg);实验结果差异性较大;不适于拷贝数过低(≤5个)或者过高菌株的分离等。因为通过该方法获得的样本信息量太少或者指纹谱过于复杂,因而不能精准区分结核分枝杆菌复合物中的菌株[11-12]。Liebana等[13]对339株结核分枝杆菌进行IS6110-RFLP分析,高达35株未见IS6110序列PCR条谱,使鉴定难以进行。

表1 插入序列IS6110和IS1081的基本信息
2间隔区寡核苷酸基因分型
1996年,人们开始应用间隔区寡核苷酸基因分型(Spoligotyping)方法对IS6110低拷贝数的结核菌进行分型研究[14]。Hermans等[15]在研究减毒牛型结核杆菌(Mycobacteriumbovis)制成的卡介苗菌株(BacillusCalmette-Guérin,BCG)时发现其基因组内存在多重直接可变重复序列(Direct variable repeat sequence,DVRS),这个区域包含保守的36 bp DNA直接重复序列(DR)和35~41 bp的间隔区(spacer)[16-17]。直接重复区域多数为成簇的规律间隔的短回文重复序列(Clustered regularly interspaced short palindromic repeats,CRISPRs),存在于所有的结核分枝杆菌复合群中,可达50个拷贝。目前,DVRS含有94个特异性的间隔区[18],间隔序列的存在与缺失可以作为分子流行病学分析的标记物。Spoligotyping主要对43个位于直接重复区域中间隔序列的存在或缺失进行划分,将合成的43个间隔序列寡核苷酸探针按顺序固定于杂交膜上,与待测PCR产物进行杂交,洗膜后用化学发光染料进行检测[19]。Spoligotyping分型商业化试剂盒较多,整个检测过程数小时即可完成,并可与Spoligotyping分型数据库SploDB4及SITVIT-WEB国际多位点遗传标志物数据库[20](收集了来自153个国家的结核分型数据,仅Spoligotyping亚型数据就有7 105个)中的数据进行比对分析。Liu等[21]采用Spoligotyping对“北京家族”亚型进行研究,该方法显示出其特有的优势,单独用于分型鉴定时分辨率达到11.90%(低于12位点VNTR的99.84%),当与VNTR分型技术联合使用时,分辨率高达99.89%。目前Spoligotyping分型与IS6110-RFLP或MLVA的组合已经成为许多国家结核分枝杆菌基因分型的基本手段之一[22]。
3多位点数目可变串联重复序列基因分型技术
在高等真核生物进化及种群的遗传进化研究中,小卫星DNA又称可变数目串联重复序列(Variable number tandem repeats,VNTR)和微卫星DNA(Microsatellite DNA)都是重要的研究工具[23]。结核分枝杆菌基因组存在很多散在分布的重复单位(Mycobacterial interspersed repetitive units,MIRU),这些是结核分枝杆菌复合群的多位点串联重复序列,多数长度为51~77 bp。MIRU以串联形式散在分布于结核分枝杆菌复合群基因组中,其重复序列的拷贝数在不同菌株中存在差异,根据这一特点可以对结核分枝杆菌进行分型。近年,结核分枝杆菌基因序列(Mycobacterium tuberculosis,Mtub),精确串联重复序列(Exact Tandem Repeats,ETR)和贝尔法斯特奎恩大学序列(Queen’s University Belfast,QUB)也较多的应用于基因分型技术,三者也是VNTR的一种形式,含有不同拷贝数形态的串联重复序列,3种序列存在重叠现象(如MIRU 4与ETR D,MIRU 31与ETR E,MIRU 27与QUB-5)。该方法对每株结核分枝杆菌复合群鉴定,并采用BioNumerics系列软件进行等位基因多样性、遗传距离和基因簇的分类以便于各个研究组间进行比较。此方法在辨别IS6110拷贝数很低(<6)的分离株时非常有效,且分辨率要高于IS6110-RFLP和Spoligotyping单独分型[24]。随着MIRU-VNTR基因分型所用重复序列的增多,分型能力也逐渐增强。Alonso-Rodriguez等[25]研究发现15个片段的MIRU-VNTR分型较12个片段分型效果有所提高,分辨率由97.8%提高至99.5%,成簇率由57.5%降至34.4%。目前,发现的41个高度保守的MIRU位点中,12个位点具有重复序列数目多态性,可以用于基因分型点[26](http://minisatellites.u-psud.fr/)。分型位点的组合方式很多,多含有12个MIRU位点,6个ETR位点,12个QUB位点和13个Mtub位点[4, 27-30]。国际上比较公认分辨力最高的3种VNTR位点组合是最早的12位点、欧洲的15位点和美国推荐的24位点分型方法(表2)[31-33]。MIRU分型分为人工分型方法和自动化分型方法两类。人工分型方法是先电泳分离PCR目的产物,然后图谱软件分析得出结果或人工直接读取。该方法适合对重复单元较大的位点进行分析,对于重复单元小于40 bp位点较难进行区分,因此人工分型方法重复性较低,不利于研究组间的结果比较。自动化分型方法利用基因分析仪检测PCR产物大小,准确性高、重复性好,该方法已用于人类基因组中的短串联重复序列分析[34]。此外,自动化分型方法可进行多重PCR扩增,分别在引物的5′端用不同的荧光染料进行标记,便于后续的分析。但自动化分型方法所需仪器和试剂较昂贵,每个样本的分析成本比人工分型法高出约5倍,不利于大规模开展。鉴于这两种分析方法各自的优缺点,研究者可以根据自己选择的MLVA和实验室具备的条件进行取舍。
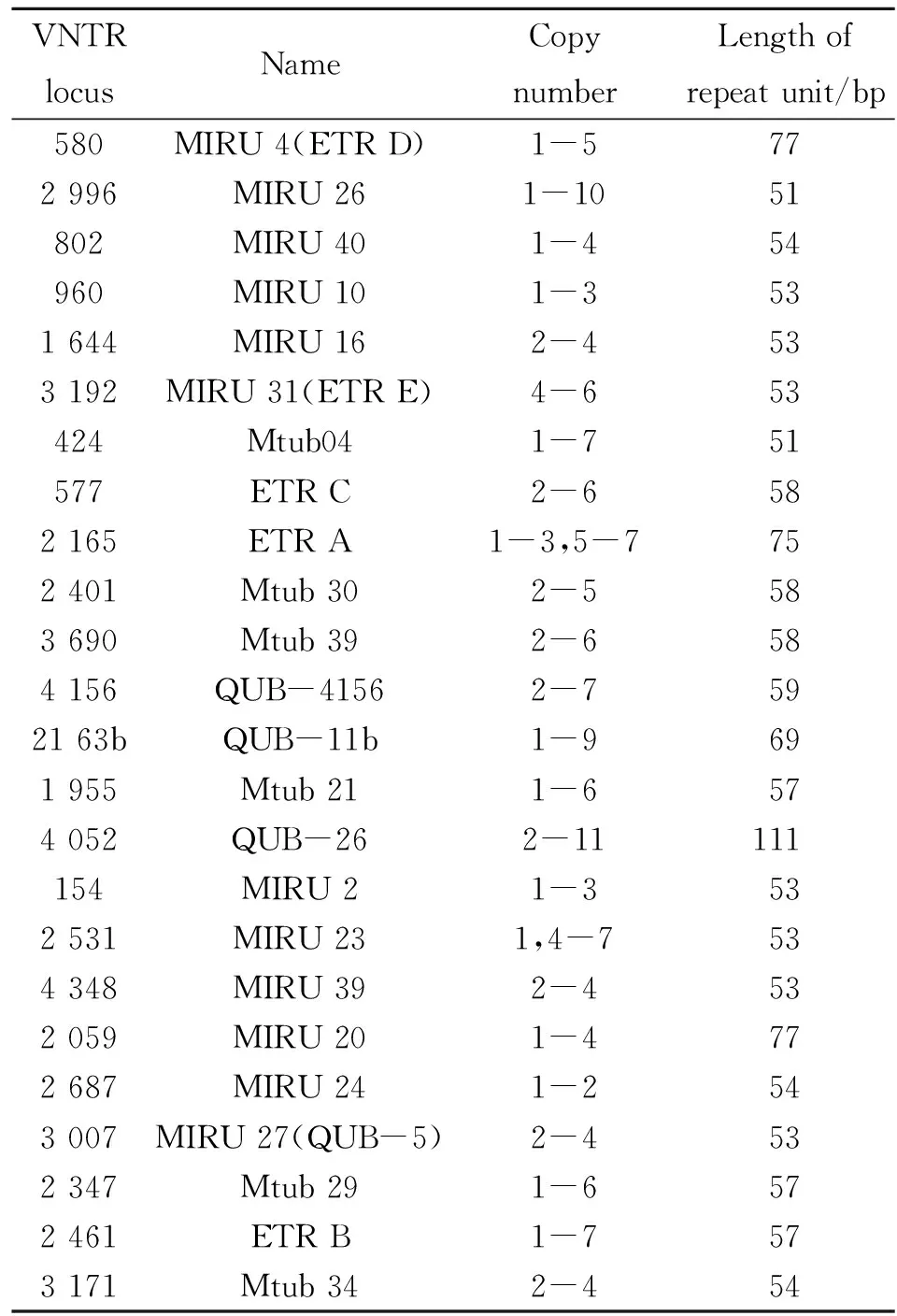
表2 MIRU-VNTR基因分型24位点信息
注:表中基因位点参照菌株H37Rv基因组比对。
Note: The reference strain is H37Rv.
4全基因组测序技术
自从20世纪70年代Sanger发明了第一代测序技术以来,测序技术得到了极快的发展,现在全基因组测序技术(Whole-genome sequencing)已广泛的应用于科研、医疗和分子流行病学等研究中,该技术的应用主要体现在4个方面:1)通过测序可以获得结核分枝杆菌的全基因组序列信息并进行菌种鉴定。Zhou等[35]利用测序得到的基因组序列进行核苷酸等长区域(1 000~10 000 bp)划分,每个区域随机核苷酸字符串(k,1 5耐药相关基因突变谱分析 随着结核分枝杆菌临床耐药株的增多,对耐药菌株进行分型从而了解其起源和流行特点显得尤为重要。近些年来,许多学者将耐药基因突变位点作为遗传标志物对结核分枝杆菌进行分型,越来越多的耐药基因突变位点被报道和研究,耐药突变谱的完善将有利于临床结核病的防治和新型抗结核药物的研发。Sreevatsan等[39]采用过氧化氢酶-过氧化物酶编码基因KatG463CTC和DNA回旋酶α亚单位编码基因gyrA95ACC作为分类依据,将MTB复合群划分为三组亚型(PGG1,PGG2,PGG3)。最近,Sengstake等[40]采用多重连接探针扩增技术(Multiplex ligation-dependent probe amplification,MLPA)和GenoTypeMTBDR plus试剂盒对耐药结核分枝杆菌进行检测,发现耐药性基因突变的发生与基因簇的划分存在联系,其是否可以作为菌株基因亚型的分子标志物还有待研究。目前对于结核耐药基因突变和其它分子标志物组合进行结核基因簇的分析尚无完全统一的标准。 6基因芯片分型技术 随着基因测序技术的发展和多个物种基因组序列测定完成,20世纪80年代末基因芯片(gene chip)技术应运而生。基因芯片(Gene-chip)又称DNA微阵列(DNA microarray),该技术是遗传学、生物化学、分子生物学等多学科的交叉融合,实现了样本检测和分析过程的连续化、集成化和微型化。结核分枝杆菌基因芯片其原理是根据结核分枝杆菌全基因组较为保守的基因序列和单核苷酸多态性位点,如Hsp65、16S rRNA、23S rRNA 、IS6110和IS1081等的保守序列设计分子探针、将其有序地固定于经过处理的载体上,然后加入标记的待测样品进行杂交,通过相应荧光检测装置获取杂交信号,来分析目的分子的有无和含量等,从而判断受检样品的菌株类型。目前已有多种结核分枝杆菌检测和分型的基因芯片应用于临床和基础研究,如耐药基因突变检测芯片、全基因组DNA测序芯片和菌种分型鉴定芯片等。Vernet等[41]研究者采用DNA微阵列技术对临床结核分枝杆菌样本进行研究,发现采用直接重复序列间隔区多态性DNA微阵列芯片(目前已经报道出94个特异的间隔区位点)存在位点差异较小的局限性,不足以对样本进行分型。而Venkataraman等[42]首次描述了应用原位合成寡核苷酸微阵列对结核分枝杆菌进行分型的方法,该微阵列包括结核分枝杆菌标准株H37Rv已知的基因和20个未注明的基因,覆盖率达到99%,经过对3862基因位点的分析,获取更为全面、特异性更强的位点信息,这对于结核分枝杆菌的流行病学研究及其未知基因的功能研究等方面具有重要的科研价值。 表3 不同分型技术之间的优缺点比较 7结语 尽管结核分枝杆菌基因分型技术近来发展迅猛,但是每种分型技术仍存在其不足及有待完善的地方,因此在实际应用中往往是采用数种组合的方案进行,现在基因芯片技术和多种分型技术的组合已经成为当今MTB分型的主流。已经基本实现了对结核病的爆发起源、变化规律、分布特点、再感染危险性以及院内感染的可能性等[43-45]的了解。Alito等[46]对结核病患者采用IS6110-RFLP指纹图谱并未发现关联,而使用Spoligotyping却解释了结核患者之间传播的内在联系。因此,新的技术出现往往会弥补前一个技术的不足。对我国来讲,由于复杂的地域环境、多样的结核病原成因及变异株家族基因簇间遗传的差异,以及每种分型技术优点和弊端(表3),完善并逐渐建立适合于我国国情的一种新标准方法,达到分辨率高、特异性好、操作方便快速,并可以提供许多的大数据库资源,是今后的发展方向,也是解决我国结核病问题的方法。 参考文献: [1]McHugh TD. World TB Day 2014: Reach the three million: a TB test, treatment and cure for all[J].Trans R Soc Trop Med Hyg, 2014, 108(3): 119-120. DOI: 10.1093/trstmh/tru006 [2]Boyle DS, McNerney R, Teng Low H, et al. Rapid detection ofMycobacteriumtuberculosisby recombinase polymerase amplification[J].PLoS One, 2014, 9(8): e103091. DOI: 10.1371/journal.pone.0103091 [3]Casart Y, Turcios L, Florez I, et al. IS6110 in oriC affects the morphology and growth ofMycobacteriumtuberculosisand attenuates virulence in mice[J].Tuberculosis(Edinb), 2008, 88(6): 545-552. DOI: 10.1016/j.tube.2008.03.006 [4]van Embden JD, Cave MD, Crawford JT, et al. Strain identification ofMycobacteriumtuberculosisby DNA fingerprinting: recommendations for a standardized methodology[J].J Clin Microbiol, 1993, 31(2): 406-409. [5]van Soolingen D, de Haas PE, Hermans PW, et al. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology ofMycobacteriumtuberculosis[J].J Clin Microbiol, 1993, 31(8): 1987-1995. [6]van Soolingen D, Hermans PW, de Haas PE, et al. Insertion element IS1081-associated restriction fragment length polymorphisms inMycobacteriumtuberculosiscomplex species: a reliable tool for recognizingMycobacteriumbovisBCG[J].J Clin Microbiol, 1992, 30(7): 1772-1777. [7]Green E, Obi LC, Okoh AI, et al. IS6110 restriction fragment length polymorphism typing of drug-resistantMycobacteriumtuberculosisstrains from northeast South Africa[J].J Health Popul Nutr, 2013, 31(1): 1-10. [8]Suffys PN, Ivens de Araujo ME, Rossetti ML, et al. Usefulness of IS6110-restriction fragment length polymorphism typing of Brazilian strains ofMycobacteriumtuberculosisand comparison with an international fingerprint database[J].Res Microbiol, 2000, 151(5): 343-351. [9]Collins DM, Hilbink F, West DM, et al. Investigation ofMycobacteriumparatuberculosisin sheep by faecal culture, DNA characterisation and the polymerase chain reaction[J].Vet Rec, 1993, 133(24): 599-600. [10]Yang ZH, de Haas PE, van Soolingen D, et al. Restriction fragment length polymorphismMycobacteriumtuberculosisstrains isolated from Greenland during 1992: evidence of tuberculosis transmission between Greenland and Denmark[J].J Clin Microbiol, 1994, 32(12): 3018-3025. [11]Kato-Maeda M, Metcalfe JZ, Flores L. Genotyping ofMycobacteriumtuberculosis: application in epidemiologic studies[J].Future Microbiol, 2011, 6(2): 203-216. DOI: 10.2217/fmb.10.165 [12]Asgharzadeh M, Kafil HS, Roudsary AA, et al. Tuberculosis transmission in Northwest of Iran: using MIRU-VNTR, ETR-VNTR and IS6110-RFLP methods[J].Infect Genet Evol, 2011, 11(1): 124-131. DOI: 10.1016/j.meegid.2010.09.013 [13]Liebana E, Aranaz A, Francis B,et al. Assessment of genetic markers for species differentiation within theMycobacteriumtuberculosiscomplex[J].J Clin Microbiol, 1996, 34(4): 933-938. [14]Aranaz A, Liebana E, Mateos A, et al. Spacer oligonucleotide typing ofMycobacteriumbovisstrains from cattle and other animals: a tool for studying epidemiology of tuberculosis[J].J Clin Microbiol, 1996, 34(11): 2734-2740. [15]Hermans PW, van Soolingen D, van Embden JD. Characterization of a major polymorphic tandem repeat inMycobacteriumtuberculosisand its potential use in the epidemiology ofMycobacteriumkansasiiandMycobacteriumgordonae[J].J Bacteriol, 1992, 174(12): 4157-4165. [16]Goyal M, Saunders NA, van Embden JD, et al. Differentiation ofMycobacteriumtuberculosisisolates by spoligotyping and IS6110 restriction fragment length polymorphism[J].J Clin Microbiol, 1997, 35(3): 647-651. [17]Driscoll JR. Spoligotyping for molecular epidemiology of theMycobacteriumtuberculosiscomplex[J].Methods Mol Biol, 2009, 551: 117-128. DOI: 10.1007/978-1-60327-999-4_10 [18]van der Zanden AG, Kremer K, Schouls LM, et al. Improvement of differentiation and interpretability of spoligotyping forMycobacteriumtuberculosiscomplex isolates by introduction of new spacer oligonucleotides[J].J Clin Microbiol, 2002, 40(12): 4628-4639. [19]Kamerbeek J, Schouls L, Kolk A, et al. Simultaneous detection and strain differentiation ofMycobacteriumtuberculosisfor diagnosis and epidemiology[J].J Clin Microbiol, 1997, 35(4): 907-914. [20]Demay C, Liens B, Burguiere T, et al. SITVITWEB--a publicly available international multimarker database for studyingMycobacteriumtuberculosisgenetic diversity and molecular epidemiology[J].Infect Genet Evol, 2012, 12(4): 755-766. DOI: 10.1016/j.meegid.2012.02.004 [21]Liu Y, Tian M, Wang XK, et al. Genotypic diversity analysis ofMycobacteriumtuberculosisstrains collected from Beijing in 2009, using spoligotyping and VNTR typing[J].PLoS One, 2014, 9(9): e106787. DOI: 10.1371/journal.pone.0106787 [22]Reves R. Universal genotyping as a tool for establishing successful partnerships for tuberculosis elimination[J].Am J Respir Crit Care Med, 2006, 174(5): 491-492. [23]Gilmore S, Peakall R, Robertson J. Short tandem repeat(STR) DNA markers are hypervariable and informative in Cannabis sativa: implications for forensic investigations[J].Forensic Sci Int, 2003, 131(1): 65-74. [24]Supply P,Allix C, Lesjean S, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing ofMycobacteriumtuberculosis[J].J Clin Microbiol, 2006, 44(12): 4498-4510. [25]Alonso-Rodriguez N, Martinez-Lirola M, Herranz M, et al. Evaluation of the new advanced 15-loci MIRU-VNTR genotyping tool inMycobacteriumtuberculosismolecular epidemiology studies[J].BMC Microbiol, 2008, 8: 34. DOI: 10.1186/1471-2180-8-34 [26]Mazars E, Lesjean S, Banuls AL, et al. High-resolution minisatellite-based typing as a portable approach to global analysis ofMycobacteriumtuberculosismolecular epidemiology[J].Proc Natl Acad Sci U S A, 2001, 98(4): 1901-1906. [27]Valcheva V, Mokrousov I, Narvskaya O, et al. Utility of new 24-locus variable-number tandem-repeat typing for discriminatingMycobacteriumtuberculosisclinical isolates collected in Bulgaria[J].J Clin Microbiol, 2008, 46(9): 3005-3011. DOI: 10.1128/JCM.00437-08 [28]Mokrousov I,Niemann S, Rastogi N. Special issue on molecular evolution, epidemiology and pathogenesis ofMycobacteriumtuberculosisand other mycobacteria[J].Infect Genet Evol, 2012, 12(4): 601. DOI: 10.1016/j.meegid.2012.02.016 [29]Arnold C. Molecular evolution ofMycobacteriumtuberculosis[J].Clin Microbiol Infect, 2007, 13(2): 120-128. [30]Kremer K, Arnold C, Cataldi A, et al. Discriminatory power and reproducibility of novel DNA typing methods forMycobacteriumtuberculosiscomplex strains[J].J Clin Microbiol, 2005, 43(11): 5628-5638. [31]Gibson A, Brown T, Baker L, et al. Can 15-locus mycobacterial interspersed repetitive unit-variable-number tandem repeat analysis provide insight into the evolution ofMycobacteriumtuberculosis[J].Appl Environ Microbiol, 2005, 71(12): 8207-8213. [32]Feiler C, Fisher AC, Boock JT, et al. Directed evolution ofMycobacteriumtuberculosisbeta-lactamase reveals gatekeeper residue that regulates antibiotic resistance and catalytic efficiency[J].PLoS One, 2013, 8(9): e73123. DOI: 10.1371/journal.pone.0073123 [33]Roring S, Scott AN, Glyn Hewinson R, et al. Evaluation of variable number tandem repeat(VNTR) loci in molecular typing ofMycobacteriumbovisisolates from Ireland[J].Vet Microbiol, 2004, 101(1): 65-73. [34]Mojtabavi Naeini M, Mesrian Tanha H, Hashemzadeh Chaleshtori M, et al. Genotyping data and novel haplotype diversity of STR markers in the SLC26A4 gene region in five ethnic groups of the Iranian population[J].Genet Test Mol Biomarkers, 2014,18(12): 820-825. DOI: 10.1089/gtmb.2014.0178 [35]Zhou F, Olman V, Xu Y. Barcodes for genomes and applications[J].BMC Bioinformatics, 2008, 9: 546. DOI: 10.1186/1471-2105-9-546 [36]O'Toole RF, Johari BM, Mac Aogain M, et al. Draft genome sequence of the first isolate of extensively drug-resistantMycobacteriumtuberculosisin New Zealand[J].Genome Announc, 2014, 2(3): e00319-14. DOI: 10.1128/genomeA.00319-14 [37]Chernyaeva EN, Shulgina MV, Rotkevich MS, et al. Genome-wideMycobacteriumtuberculosisvariation(GMTV) database: a new tool for integrating sequence variations and epidemiology[J].BMC Genomics, 2014, 15: 308. DOI: 10.1186/1471-2164-15-308 [38]Gardy JL, Johnston JC, Ho Sui SJ, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak[J].N Engl J Med, 2011, 364(8): 730-739. DOI: 10.1056/NEJMoa1003176 [39]Sreevatsan S, Pan X, Stockbauer KE, et al. Restricted structural gene polymorphism in theMycobacteriumtuberculosiscomplex indicates evolutionarily recent global dissemination[J].Proc Natl Acad Sci U S A, 1997, 94(18): 9869-9874. [40]Sengstake S, Bablishvili N, Schuitema A, et al. Optimizing multiplex SNP-based data analysis for genotyping ofMycobacteriumtuberculosisisolates[J].BMC Genomics, 2014, 15: 572. DOI: 10.1186/1471-2164-15-572 [41]Vernet G, Jay C, Rodrigue M, et al. Species differentiation and antibiotic susceptibility testing with DNA microarrays[J].J Appl Microbiol, 2004, 96(1): 59-68. [42]Venkataraman B, Vasudevan M, Gupta A. A new microarray platform for whole-genome expression profiling ofMycobacteriumtuberculosis[J].J Microbiol Methods, 2014, 97: 34-43. DOI: 10.1016/j.mimet.2013.12.009 [43]Maciel EL, Prado TN, Peres RL, et al. Guided sputum sample collection and culture contamination rates in the diagnosis of pulmonary TB[J].J Bras Pneumol, 2009, 35(5): 460-463. [44]Moore DA, Caviedes L, Gilman RH, et al. Infrequent MODS TB culture cross-contamination in a high-burden resource-poor setting[J].Diagn Microbiol Infect Dis, 2006, 56(1): 35-43. [45]Sola C, Filliol I, Legrand E, et al. Genotyping of theMycobacteriumtuberculosiscomplex using MIRUs: association with VNTR and spoligotyping for molecular epidemiology and evolutionary genetics[J].Infect Genet Evol, 2003, 3(2): 125-133. [46]Alito A, Morcillo N, Scipioni S, et al. The IS6110 restriction fragment length polymorphism in particular multidrug-resistantMycobacteriumtuberculosisstrains may evolve too fast for reliable use in outbreak investigation[J].J Clin Microbiol, 1999, 37(3): 788-791. [47]McEvoy CR, Falmer AA, Gey van Pittius NC, et al. The role of IS6110 in the evolution ofMycobacteriumtuberculosis[J].Tuberculosis(Edinb), 2007, 87(5): 393-404. [48]Lari N,Rindi L, Garzelli C. Identification of one insertion site of IS6110 inMycobacteriumtuberculosisH37Ra and analysis of the RvD2 deletion inM.tuberculosisclinical isolates[J].J Med Microbiol, 2001, 50(9): 805-811. [49]Warren RM, Sampson SL, Richardson M, et al. Mapping of IS6110 flanking regions in clinical isolates ofMycobacteriumtuberculosisdemonstrates genome plasticity[J].Mol Microbiol, 2000, 37(6): 1405-1416. [50]Aga RS, Fair E, Abernethy NF, et al. Microevolution of the direct repeat locus ofMycobacteriumtuberculosisin a strain prevalent in San Francisco[J].J Clin Microbiol, 2006, 44(4): 1558-1560. [51]Warren RM, Streicher EM, Sampson SL, et al. Microevolution of the direct repeat region ofMycobacteriumtuberculosis: implications for interpretation of spoligotyping data[J].J Clin Microbiol, 2002, 40(12): 4457-4465. [52]Jamieson FB, Teatero S, Guthrie JL, et al. Whole-genome sequencing of theMycobacteriumtuberculosisManila sublineage results in less clustering and better resolution than mycobacterial interspersed repetitive-unit-variable-number tandem-repeat(MIRU-VNTR) typing and spoligotyping[J].J Clin Microbiol, 2014, 52(10): 3795-3798. DOI: 10.1128/JCM.01726-14 [53]Supply P, Mazars E, Lesjean S, et al. Variable human minisatellite-like regions in theMycobacteriumtuberculosisgenome[J].Mol Microbiol, 2000, 36(3): 762-771. [54]Alonso M, Alonso Rodriguez N, Garzelli C, et al. Characterization ofMycobacteriumtuberculosisBeijing isolates from the Mediterranean area[J].BMC Microbiol, 2010, 10: 151. DOI: 10.1186/1471-2180-10-151 [55]Colangeli R, Arcus VL, Cursons RT, et al. Whole genome sequencing ofMycobacteriumtuberculosisreveals slow growth and low mutation rates during latent infections in humans[J].PLoS One, 2014, 9(3): e91024. DOI: 10.1371/journal.pone.0091024 [56]Mulenga C, Shamputa IC, Mwakazanga D, et al. Diversity ofMycobacteriumtuberculosisgenotypes circulating in Ndola, Zambia[J].BMC Infect Dis, 2010, 10: 177. DOI: 10.1186/1471-2334-10-177 [57]Smittipat N, Billamas P, Palittapongarnpim M, et al. Polymorphism of variable-number tandem repeats at multiple loci inMycobacteriumtuberculosis[J].J Clin Microbiol, 2005, 43(10): 5034-5043. [58]Pang Y, Xia H, Zhang ZY, et al. Multicenter evaluation of genechip for detection of multidrug-resistantMycobacteriumtuberculosis[J].J Clin Microbiol, 2013, 51(6): 1707-1713. DOI: 10.1128/JCM.03436-12 [59]Zhang ZH, Li LT, Luo F, et al. Rapid and accurate detection of RMP- and INH- resistantMycobacteriumtuberculosisin spinal tuberculosis specimens by CapitalBio DNA microarray: a prospective validation study[J].BMC Infect Dis, 2012, 12: 303. DOI: 10.1186/1471-2334-12-303 DOI:10.3969/j.issn.1002-2694.2016.01.016 通讯作者:张阿梅,Email: zam1980@yeah.net 中图分类号:R378.91 文献标识码:A 文章编号:1002-2694(2016)01-0076-07 Corresponding author:Zhang A-mei, Email: zam1980@yeah.net 收稿日期:2015-05-13;修回日期:2015-11-05 Genetic genotyping techniques for Mycobacterium tuberculosis and the application LI Dao-qun1,2,XIA Xue-shan1,2,SONG Yu-zhu1,2,ZHANG A-mei1,2 (1.FacultyofLifeScienceandTechnology,KunmingUniversityofScienceandTechnology,Kunming650500,China;2.InstituteofMolecularMedicine,KunmingUniversityofScienceandTechnology,Kunming650500,China) Abstract:Mycobacterium tuberculosis (MTB) is the pathogenic agent for TB. Recently, the incidence rate of TB and the TB strains with drug resistant increased. All suggested that the epidemiological characters of TB were different from previous one. Choosing suitable therapeutic methods could efficiently protect normal individuals from TB and treat the patients with TB, which is essential for personalized medicine and treatment. The common genotyping methods included restriction fragment length polymorphism of IS6110, spacer oligonucleotide typing, multi-locus variable tandem number repeats analysis, whole-genome sequencing, resistance-associated mutation spectrum analysis, microarray genotyping, etc. Two sides, i.e. advantage and shortcoming, co-existed in each technology when they were used in clinic and research. The genotyping methods and the application were summarized in this review to provide theoretical basis for clinicians and researchers. Keywords:Mycobacterium tuberculosis; typing technology; genotyping 云南省科技厅重点新产品开发计划 (No.2012AE003和No.2013BC009),昆明理工大学人才培养项目(No.KKSY201226148)资助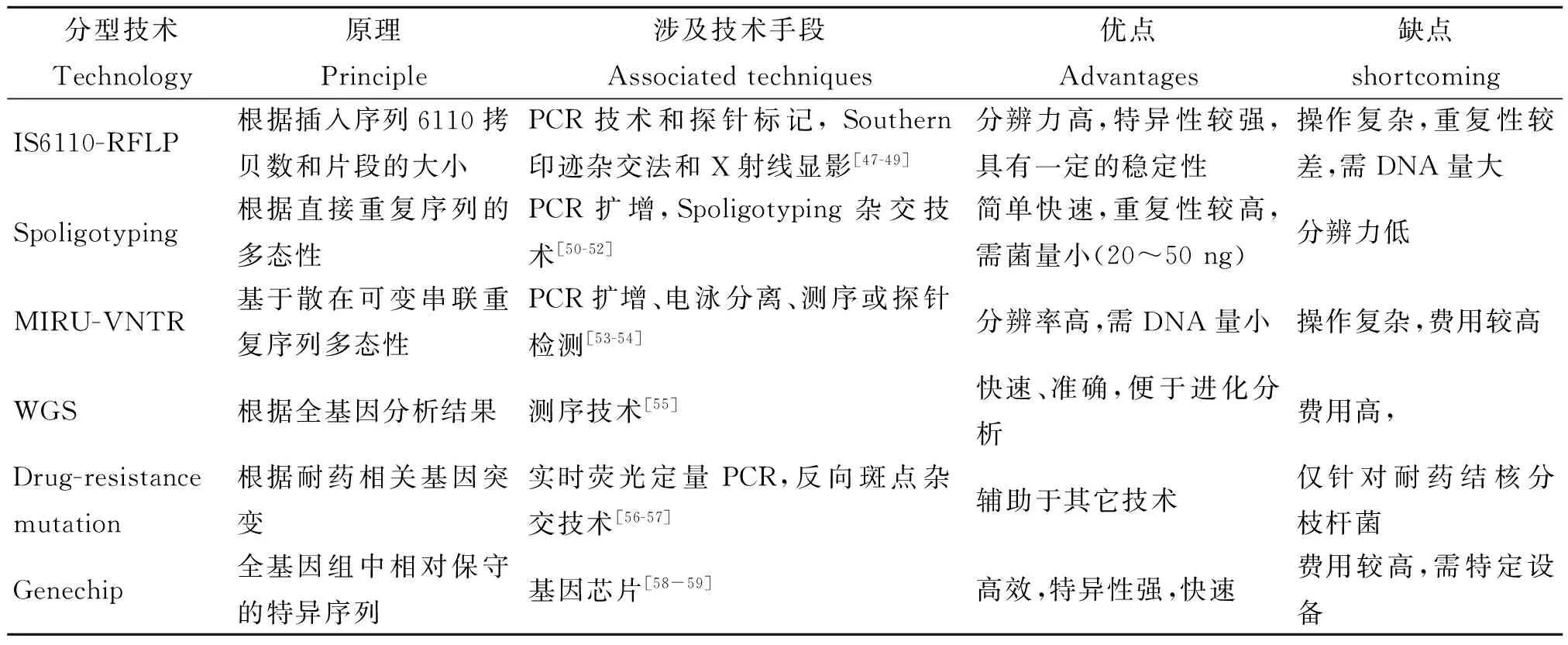
猜你喜欢
糖尿病新世界(2017年2期)2017-05-17 12:43:30
安徽农学通报(2017年8期)2017-05-12 10:40:45
上海医药(2017年5期)2017-04-14 05:31:14
江苏农业科学(2016年7期)2016-10-20 01:13:21
中国实用医药(2016年18期)2016-08-03 09:49:34
中国实用医药(2016年9期)2016-05-17 10:59:06
中外医疗(2015年3期)2015-08-29 20:31:50
医学信息(2015年14期)2015-04-29 17:36:37
中国社区医师(2014年22期)2015-04-16 21:44:21
中国民族民间医药·下半月(2014年6期)2015-01-21 06:24:29
