
离子液体辅助合成磁性介孔炭微球及其对萘普生释放的影响
2016-07-27 00:54宋镠黄皑琳鲁泊宏邱江龙潘柔君王秀芳
广东药科大学学报 2016年3期
关键词:合成
宋镠,黄皑琳,鲁泊宏,邱江龙,潘柔君,王秀芳
离子液体辅助合成磁性介孔炭微球及其对萘普生释放的影响
宋镠,黄皑琳,鲁泊宏,邱江龙,潘柔君,王秀芳
(广东药科大学药学院,广东 广州510006)
摘要:目的考察磁性介孔炭对萘普生的吸附与释放行为。方法通过简单的原位一锅法,以绿色溶剂离子液体辅助合成磁性介孔炭微球,通过N2吸附SEM、大角XRD和拉曼光谱对磁性介孔炭微球进行表征;并考察不同pH条件下,磁性介孔炭微球对萘普生释放的影响。结果随着Fe(NO3)3·9H2O用量的增大,磁性介孔炭微球的比表面积、孔径及孔容逐渐减小。磁性介孔炭微球大大提高了萘普生的释放速率,且累积释放率随着Fe-C的比表面积和孔容的增大而增大。在所研究的pH范围内(5.0,6.8,8.0),当pH=8.0时,萘普生在Fe-C-1上的载药量最高、释放最快,且累积释放速率最大。结论萘普生在磁性介孔炭微球上的释放分为3个阶段:第1阶段为中孔扩散,表面扩散过程占主导地位;第2阶段为孔隙扩散速率限制阶段,是萘普生被吸附进炭微球的内部毛孔后再逐渐释放的过程;第3阶段为释放平衡状态。
关键词:磁性介孔炭微球;合成;离子液体;萘普生
网络出版时间:2016-05-23 10:59 网络出版地址:http://www.cnki.net/kcms/detail/44.1413.R.20160523.1059.001.html
近年来,介孔炭微球材料因具有高比表面、热稳定性、生物相容性好和化学惰性等优点,得到越来越多的关注[1],在吸附、药物载体、分离催化和能源存储系统得到广泛应用。但是,传统合成的介孔炭材料由于粒径较小,难以从溶液中分离。研究人员通常通过引入磁性源来解决这一缺点,如Wang等[2]利用铁纳米材料合成铁磁性介孔炭材料,使介孔材料易于从溶液中分离。此外,许多合成过程中均会使用挥发性溶剂作为反应介质,如Fang等[3]使用氨水和甲醛溶液,Zhu等[4]使用HCl来合成介孔炭。但是,挥发性溶剂存在环境污染、腐蚀设备和危害人体健康等问题。因此,需要一种环境友好型的溶剂来合成介孔炭材料。有研究报道,水热法可以成功合成介孔炭微球[5]。但是,水热法需要严格的制备步骤,包括高温和压力,在溶剂处理环节中,炭源不能在沉淀中完全转化为炭微球,导致产率较低。
萘普生(naproxen,Nap)又名(S)-(+)-2-(6-甲氧基-2-萘基)丙酸,是一种非甾体抗炎药,广泛用于治疗关节炎、术后疼痛、头痛和肌肉疼痛骨骼[6]。萘普生的溶解度低,在胃肠道中难以吸收,生物利用度差,而介孔炭材料作为药物载体可以有效地控制药物分子的增溶和释放速率,同时能够保持药物结构的完整性。
本研究拟通过简单的原位一锅法,以绿色溶剂离子液体辅助合成磁性介孔炭微球,所有的试剂都是一次性加入,方法简单、易操作、原料廉价,特别是不使用挥发性溶剂,从源头消除对环境造成的污染。在该方法中,炭源在溶剂蒸发的过程中完全转化为炭微球。同时,选用萘普生为模型药物,考察所合成的磁性介孔炭微球在不同pH下对萘普生的吸附与释放行为,旨在为磁性介孔炭微球对萘普生药物的可控释放提供理论依据。
1 材料与方法
1.1 原料与仪器
萘普生(分析纯,大连美仑有限公司);溴代-1-丁基-3-甲基咪唑(分析纯,上海紫一试剂厂);糠醇(分析纯,上海金山亭新化工试剂厂);其余试剂均为分析纯。
X-射线衍射仪(荷兰PANalytical X'Pert Pro);Tristar 3020扫描电子显微镜(Micrometrics,USA);ZRS-6G溶出度仪(德国 LEO 1530VP);752N紫外可见分光光度计(上海精密科学仪器公司);KTL-140管式炉(南京大学仪器厂)。
1.2 磁性介孔炭微球的合成
以糠醇为炭源,Fe(NO3)3·9H2O为磁性前躯体,SiO2纳米微粒为致孔剂,离子液体溴代-1-丁基-3-甲基咪唑([C4mim]Br)辅助合成磁性介孔炭微球:将1.2 mmol的Fe(NO3)3·9H2O加入0.5 mL [C4mim]Br中,再加入糠醇2 mL和SiO21 g,在室温下超声10 min后,直接放入管式炉,在N2中,先以80℃保持6 h,再升温至850℃保持3 h。将产物用NaOH-乙醇水溶液(1 mol/L)洗去SiO2微球,重复4次,用去离子水清洗至pH=7,在真空干燥箱中40℃干燥得磁性介孔炭微球,所合成的材料命名为Fe-C-1。Fe-C-2和Fe-C-3的合成同上述步骤,仅改变Fe(NO3)3·9H2O摩尔量分别为2.0 mmol和2.5 mmol。
1.3 药物加载
奈普生溶解在10 mL乙醇中,萘普生与磁性介孔炭微球质量比为1∶1,磁力搅拌3 h,分别加载到磁性介孔炭微球Fe-C-1、Fe-C-2和Fe-C-3中。载药后的样品在80℃下烘干除去溶剂,载药后的样品分别命名为Nap-Fe-C-1、Nap-Fe-C-2和Nap-Fe-C-3。
1.4 萘普生释放试验
萘普生药物释放试验按照《中国药典》2015年版二部中的桨法进行:温度(37.0±0.5)℃,转速为100 r/min,以pH 5.0、6.8、8.0磷酸盐缓冲溶液900 mL为溶剂,在5、10、15、20、25、30、45、60、75、90、120、150、180 min时分别取溶液10 mL,微孔滤膜滤过,并同时补充同温等量溶剂,取上述续滤液在331 nm处测定吸光度,计算在不同时间的累积释放率。
1.5 表征
比表面积和孔径分布分别采用 Brunauer-Emmett-Teller(BET)和Barett-Joyner-Halenda(BJH)方法获得;总孔容根据相对压力P/P0为0.99时的吸附量计算。
2 结果与分析
炭微球Fe-C-1、Fe-C-2和Fe-C-3的吸附和脱吸附等温线均属于典型的Ⅳ等温线,在相对压力P/P0为0.5~0.9范围内有1个明显的滞后回环(图1A),这是介孔材料的毛细管凝聚现象,是介孔结构形成的特征。磁性介孔炭微球的孔结构参数见表1。可见,Fe-C-1微球显示出最大的比表面积(653.4 m2/g)和最大的孔容(1.27 cm3/g)。磁性介孔炭微球从Fe-C-1 到Fe-C-3的孔径从9.3 nm下降到6.6 nm(见图1B),这是由于随着Fe(NO3)3·9H2O用量逐渐变大,铁磁性纳米微粒密度增大,微球结构部分不完整,导致材料比表面积和孔容大小随着Fe(NO3)3· 9H2O用量增大而减小。此外,萘普生的分子尺寸为1.172 nm×0.501 nm[7],明显小于所合成炭微球Fe-C的孔径(6~9 nm),因此能够载入到磁性介孔炭微球的孔道内。
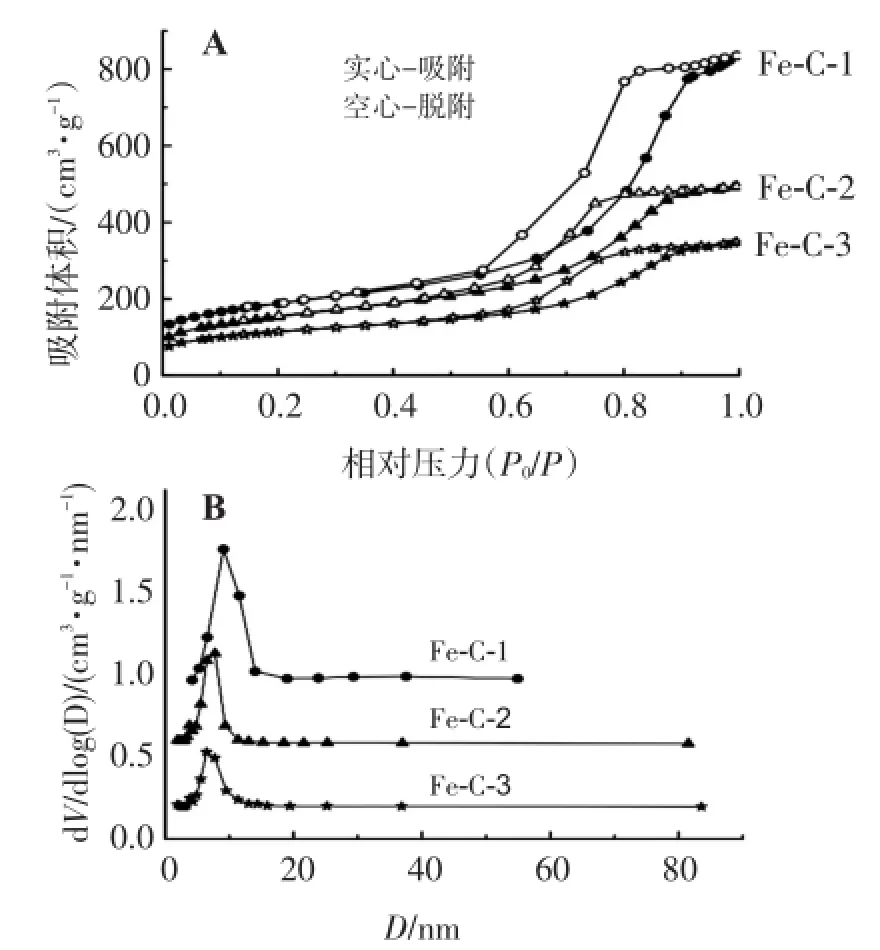
图1 磁性介孔炭微球的N2吸附、脱吸附等温线(A)和孔径分布曲线(B)Figure 1 Nitrogen adsorption isotherms(A)and pore size distributions(B)of the materials
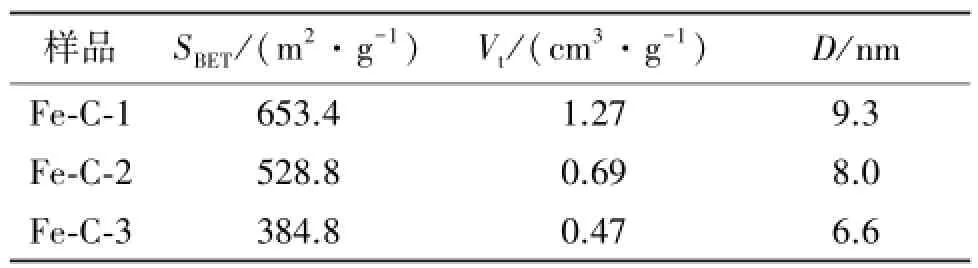
表1 磁性介孔炭微球的孔结构参数Table 1 The pore structure parameters of magnetic mesoporous carbon microspheres
Fe-C-1、Fe-C-2和Fe-C-3的SEM图见图2。可见,3种炭材料均为球形结构,粒径在15~20 μm,随着Fe(NO3)3·9H2O用量增多,炭微球完整性下降。

图2 磁性介孔炭微球的SEM图Figure 2 The SEM images of the magnetic mesoporous carbon microspheres
磁性介孔炭微球Fe-C-1、Fe-C-2和Fe-C-3材料的大角X射线衍射图见图3。可见,3种材料均在44.6°有1个强的(110)衍射峰,为体心立方结构,是α-Fe(JCPDS card No.06-0696)的衍射峰,α-Fe可能是通过Fe2O3或Fe(NO3)3在850℃的炭化过程中还原反应形成的。Fe-C-1、Fe-C-2在26.3°中有1个较弱的衍射峰,Fe-C-3则表现为1个较强的衍射峰,表明随着Fe(NO3)3·9H2O用量增加利于石墨化进程。26.3°处为(002)的衍射峰,是石墨炭的特征衍射峰(JCPDS card No.008-0415)。根据晶面的最大衍射角(θ值)算出(002)衍射面间距(d-spacing)为0.340 nm,比石墨的衍射面间距(0.335 nm)略大[8],表明磁性介孔炭微球在850℃的焙烧炭化过程中被石墨化了,比纯炭的石墨化温度(2 400℃)低很多[9],这是因为在惰性气体环境中,炭材料和磁性金属在共同的高温焙烧过程中,加速了石墨炭结构的生成,而且铁纳米微粒具有催化作用,使磁性介孔炭微球具有比较低的石墨化温度[10-11]。
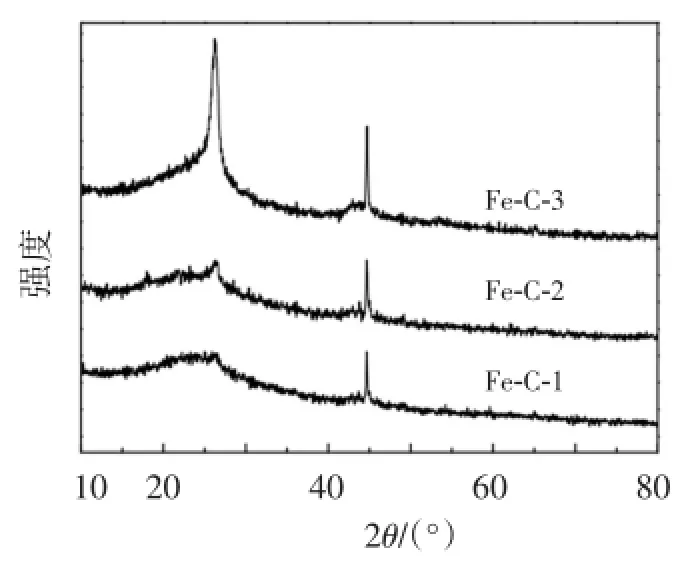
图3 磁性介孔炭微球的大角XRD衍射图谱Figure 3 Wide-angle X-ray diffraction(XRD)patterns of magnetic mesoporous carbon microspheres
磁性介孔炭微球的拉曼光谱图见图4。可见,在1 590 cm-1(G带)处有1个石墨炭的C-C之间sp2伸缩振动峰,G带强而窄的峰形表明了石墨炭的存在,与XRD衍射图的结果相互印证。此外,在1 330 cm-1(D带)处有1个无定形结构,表明石墨炭的结构有一定的缺陷。随着Fe(NO3)3·9H2O用量的增加,Fe-C-1、Fe-C-2和 Fe-C-3的 ID/IG分别为2.46、1.68和1.08,可知结晶石墨化程度增加。这与磁性介孔炭微球的大角XRD结果一致。
萘普生纯药和Nap-Fe-C在pH 6.8磷酸盐缓冲溶液中的累积释放曲线见图5。可见,pH=6.8时,萘普生纯药的释放最慢,90 min累积释放量为60.8%,Nap-Fe-C-1、Nap-Fe-C-2和Nap-Fe-C-3的累积释放率分别为85.8%、80.6%和73.7%。结果表明,磁性介孔炭微球Fe-C大大提高了萘普生的释放,而且累积释放率随着Fe-C的比表面积和孔容的增大而增大。这是由于比表面积较大的Fe-C可以吸附更多的药物并且与溶出介质有更多的接触,从而提高了萘普生的溶解速度。药物释放表现出类似的释放曲线,可以分为3个阶段:第1阶段为中孔扩散,表面扩散过程占主导地位;第2阶段为孔隙扩散速率限制阶段,是萘普生被吸附进炭微球的内部毛孔后再逐渐释放的过程;第3阶段为平衡状态[12]。

图4 磁性介孔炭微球的拉曼光图谱Figure 4 Raman spectra of the synthesized magnetic mesoporous carbon spheres
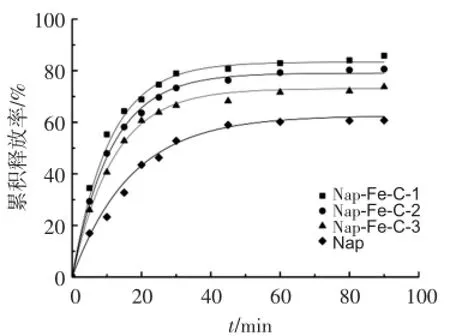
图5 萘普生、Nap-Fe-C-1、Nap-Fe-C-2和Nap-Fe-C-3的释放曲线(pH=6.8)Figure 5 The release curves of naproxen,Nap-Fe-C-1,Nap-Fe-C-2 and Nap-Fe-C-3(pH=6.8)
为了深入研究磁性介孔炭微球对药物的释放机理,采用以下指数方程对药物释放进行拟合[13]:
y=a(1-e-kt),
式中,y为药物的累积释放速率,a为萘普生的最大累积释放量,K为速率常数,t为药物释放时间。拟合参数见表2,可见Nap-Fe-C-1、Nap-Fe-C-2、Nap-Fe-C-3的载药量分别为76.1%、73.8%和58.3%,这个趋势与炭微球的比表面积大小一致。由于炭微球的比表面积及孔容越大,有更多空隙和容积容纳萘普生,提高材料载药率。a值和k值从大到小的顺序是Nap-Fe-C-1、Nap-Fe-C-2、Nap-Fe-C-3,这个趋势与炭微球的比表面积和孔容大小相一致。这表明萘普生载入Fe-C后,磁性炭微球限制了萘普生的重结晶,使它保持无定形结构。此外,3种磁性炭微球的累积释放率均没有达到100%,这是由于萘普生分子与磁性炭微球之间存在的疏水作用阻碍了萘普生的释放。

表2 磁性介孔炭微球的Nap-Fe-C拟合参数(pH=6.8)Table 2 The fitting parameters of magnetic mesoporous carbon spheres Nap-Fe-C(pH=6.8)
Nap-Fe-C-1在不同pH磷酸盐缓冲溶液下萘普生的累积释放率曲线见图6。可见,释放速率快慢顺序是pH 8.0>pH 6.8>pH 5.0,原因是萘普生的Ka(pKa=4.2)值是一个定值,当溶液中pH变大时,[H+]浓度下降[A-]浓度升高(见表3),药物的溶解度增加,累积释放率提高。因此,药物在酸性溶液中的累积释放速率低于碱性溶液。

图6 Nap-Fe-C-1在不同pH磷酸盐缓冲溶液的释放行为Figure 6 The release behavior of Nap-Fe-C-1 under differentpH value
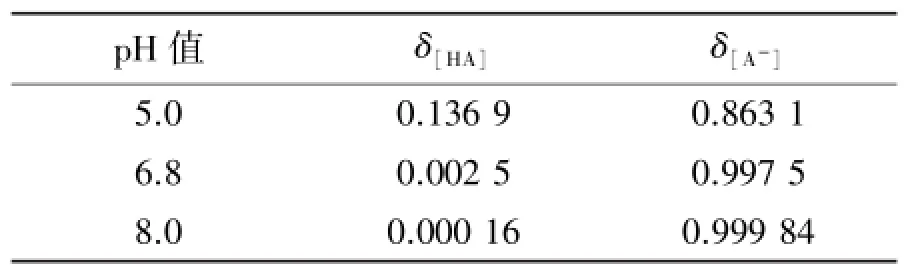
表3 萘普生在不同pH下的分布系数Table 3 The distribution coefficient of Naproxen under different pH value
3 结论
通过原位一锅法简单快捷合成磁性介孔炭微球,随着Fe(NO3)3·9H2O用量的增大,磁性介孔炭微球的比表面积、孔径及孔容逐渐减小。当 Fe (NO3)3·9H2O用量最小时,磁性介孔炭微球Fe-C-1的比表面积、孔径及孔容最大,对萘普生的载药量最高、释放最快。萘普生在pH8.0磷酸盐缓冲溶液的累积释放速率最高。萘普生在磁性介孔炭微球上的累积释放可以分为3个阶段:第1阶段为中孔扩散,意味着表面扩散过程占主导地位;第2阶段为孔隙扩散速率限制阶段,是萘普生吸附进炭微球的内部毛孔后再逐渐释放的过程;第3阶段为释放平衡状态。
参考文献:
[1]LIU Jian,WICKRAMARATNE N P.Molecular-based design and emerging applications of nanoporous carbon spheres [J].Nat Mater,2015,14(8):763-774.
[2]WANG Xiqing,LIANG Chengdu,DAI Sheng.Facial synthesis of ordered mesoporous carbons with high thermal stability by self-assemblyofresorcinol-formaldehydeandblock copolymers under highly acidic conditions[J].Langmuir,2008,24(14):7500-7505.
[3]FANG Yin,GU Dong,ZOU Ying,et al.A low-concentration hydrothermal synthesis of biocompatible ordered mesoporous carbon nanospheres with tunable and uniform size[J]. Angew Chem Int Ed,2010,49(43):7987-7991.
[4]ZHU Wenquan,WAN Long,ZHANG Chen,et al.Exploitation of 3D face-centered cubic mesoporous silica as a carrier for a poorly water soluble drug:influence of pore size on release rate[J].Mater Sci Eng C,2014,34:78-85.
[5]WANG Qing,LI Hong,CHEN Liquan,et al.Monodispersed hard carbon spherules with uniform nanopores[J].Carbon, 2001,39(14):2211-2214.
[6]ROSTOM A,MUIR K,DUBE C,et al.Prevention of NSAID-related uppergastrointestinaltoxicity:ametaanalysis of traditional NSAIDs with gastroprotection and COX-2 inhibitors[J].Drug Healthcare Patient Saf,2009 (1):47-71.
[7]CARRIAZO D,ARCO M D,MARTIN C,et al.Influence of the inorganic matrix nature on the sustained release of naproxen[J].Microporous Mesoporous Mater,2010,130:229-238.
[8]HIRAOKA T,KAWAKUBO T,KIMURA J,et al.Selective synthesis of double-wall carbon nanotubes by CCVD of acetylene using zeolite supports[J].Chem Phys Lett,2003,382:679-685.
[9]WEISWEILER W,SUBRAMANIAN N,TERWIESCH B. Catalytic influence of metal melts on the graphitization of monolithic glasslike carbon[J].Carbon,1971,9:755-761.
[10]YAO J,LI L,SONG H,et al.Synthesis of magnetically separable ordered mesoporous carbons from F127/[Ni (H2O)6](NO3)2/resorcinol-formaldehyde composites[J]. Carbon,2009,47:436-444.
[11]LI J,GU J,LI H,et al.Synthesis of highly ordered Fecontainingmesoporouscarbonmaterialsusingsoft templating routes[J].Microporous Mesoporous Mater,2010,128:144-149.
[12]VALDERRAMA C,GAMWASANS X,HERAS D L,et al. Sorption kinetics ofpolycyclicaromatichydrocarbons removal usinggranularactivatedcarbon:intraparticle diffusion coefficients[J].J Hazard Mater,2008,157(2):386-396.
[13]BALAS F,MANZANO M,COLILLA M,et al.L-Trp adsorption into silica mesoporous materials to promote bone formation[J].Acta Biomater,2008,4(3):514-522.
(责任编辑:陈翔)
中图分类号:R914;O647.3
文献标志码:A
文章编号:1006-8783(2016)03-0295-05
DOI:10.16809/j.cnki.1006-8783.2016030402
收稿日期:2016-03-04
基金项目:国家级大学生创新创业训练计划项目(201510573002);国家自然科学基金项目(21373061);广东省自然科学基金重点项目(2014A030311038)
作者简介:宋镠(1995—),男,药学专业2013级本科生;通信作者:王秀芳(1977—),女,博士,教授,主要从事功能炭材料研究,Email:x_f_wang@163.com。
Ionic liquids assisted synthesis of magnetic mesoporous carbon microspheres and its effect on the release of naproxen
SONG Liu,HUANG Ailin,LU Bohong,QIU Jianglong,PAN Roujun,WANG Xiufang
(School of Pharmacy,Guangdong Pharmaceutical University,Guangzhou 510006,China)
Abstract:Objective To study the adsorption and release behavior of naproxen on carbon spheres.
Methods Magnetic mesoporous carbon spheres were synthesized via a simple in-situ one pot method assisted by a green solvent ionic liquid.The magnetic mesoporous carbon spheres were characterized by N2adsorption isotherms,SEM,wide-angle XRD and Raman spectra.The impact of pH value on the release rate of naproxen was estimated.Results The addition of Fe(NO3)3·9H2O increased with the decrease of the specific surface area,pore diameter and pore volume of the materials.Within the pH range of this study (5.0,6.8,8.0),Fe-C-1 exhibited the largest loading amount of naproxen,the fastest release rate and cumulative release rate at pH 8.0.Conclusions The cumulative release of naproxen in magnetic mesoporous carbon microspheres could be split into three stages.The first stage was the mesopore diffusion,in which surface diffusion dominated the process.The second stage was the pore diffusion rate limiting stage,in which naproxen was adsorbed into the pores of carbon microspheres,and then was gradually released.The last stage was the balance state of the drug release.
Key words:mesoporous carbons spheres;synthesis;ionic liquids;naproxen
猜你喜欢
现代商贸工业(2016年14期)2016-12-27
安徽理工大学学报·自然科学版(2016年4期)2016-12-23
考试周刊(2016年85期)2016-11-11
农业与技术(2016年15期)2016-11-09
科技视界(2016年18期)2016-11-03
科教导刊·电子版(2016年19期)2016-08-19
科技视界(2016年15期)2016-06-30
科技视界(2016年10期)2016-04-26
科技视界(2016年9期)2016-04-26
科技视界(2016年3期)2016-02-26
