
双金属氰化络合催化剂催化环氧烷烃与二氧化碳共聚研究进展
2016-07-26 09:52张鹏刘定华南京工业大学化学化工学院江苏南京210009
化工进展 2016年7期
关键词:二氧化碳
张鹏,刘定华(南京工业大学化学化工学院,江苏 南京 210009)
综述与专论
双金属氰化络合催化剂催化环氧烷烃与二氧化碳共聚研究进展
张鹏,刘定华
(南京工业大学化学化工学院,江苏 南京 210009)
摘要:针对 CO2与环氧烷烃共聚制备脂肪族聚碳酸酯反应体系,本文在综合分析脂肪族聚碳酸酯应用前景的基础上,着重介绍了双金属氰化络合催化剂在该工艺中的研究进展,并对不同种类活性中心金属的组合在不同反应体系中的催化效果进行了分析。共沉淀法制得的Zn-Co双金属氰化络合催化剂在CO2与环氧烷烃的催化体系中,通过改变制备方法、配体等条件可以实现对催化剂形貌的控制,有利于催化活性的提升。Zn-Fe双金属氰化络合催化剂对于环氧丙烷开环活性较好。同时还介绍了采用其他活性中心金属组合制得的双金属氰化络合催化剂,发现对于CO2与环氧烷烃共聚活性受中心金属及配体的影响较大。最后,对该共聚反应的机理进行了分析,为双金属氰化络合催化剂的设计研究指明了方向。
关键词:双金属络合催化剂;二氧化碳;反应机理;脂肪族聚碳酸酯
鉴于石油资源将在未来100年内消耗殆尽,高效地利用可再生资源吸引了越来越多的关注[1]。CO2作为一种潜在的可再生C1资源,具有无毒、不燃、来源广泛的特点,高效地利用CO2不仅能够从一定程度上缓解日益严重的能源危机,而且可以解决温室气体过度排放带来的全球温度上升的问题[2-5]。但是CO2高度的热力学稳定性限制了其广泛应用。为了克服CO2自身存在的不足,大量的研究将其与高活性的物质进行反应,在过去的半个世纪里,CO2和杂环化合物催化耦合吸引了广泛的关注[6-13]。其中大量文献报道了CO2与环氧烷烃合成脂肪族聚碳酸酯或环状碳酸酯。
脂肪族聚碳酸酯(PPC)相较于芳香族聚碳酸酯具有更低的玻璃化转化温度Tg[14]、更好的抗拉强度和弹性模量,同时PPC的介电性能良好且对气体的通过率较低,这使得PPC可制成薄膜应用于食品保鲜领域[15-16]。PPC极易水解,CO2-EO(环氧乙烷)的共聚物具有生物降解性,植入动物体内一周左右即可以降解完全。其他的共聚物也基本都能与生物体良好相容,可被微生物彻底分解而不留残渣,这可以缓解由塑料的不可降解性引发的“白色污染”问题。在PPC中引入缩水甘油醚衍生物等可水解基团后,能应用于药物缓释剂以及其他医疗材料中[17-20]。通过对聚合条件的控制,可以得到具有预定相对分子质量和羟基官能团的PPC树脂,作为聚氨酯的合成原料,可以制得耐水解的聚碳酸酯型聚氨 酯[21-23]。通过引入第三种单体进行共聚,可以得到含碳酸酯基和酯基的不饱和树脂,通过交联固化后,可以得到一种能代替普通不饱和聚酯使用的新材料[24]。
总之,随着PPC合成技术的不断进步,PPC的应用领域将越来越广。因而高效合成PPC成为目前的一个热点,而高效合成的关键就在于催化剂的开发。本文在综合分析PPC合成进展的基础上,着重介绍双金属氰化络合催化剂的研究现状,旨在探讨双金属氰化络合催化剂的开发思路和研究方向。
1 双金属氰化络合催化剂研究现状
双金属氰化络合物(double metal cyanide complex,DMC)催化剂是一种首先应用环氧丙烷(PO)开环聚合制备高相对分子质量聚醚化合物的高效催化剂,1974年由美国通用橡胶公司首先报道出来[25],一般由水溶性的金属氰化络合物和金属化合物以及合适的有机配体反应制得,典型的 DMC催化剂组成式为MII[M(CN)n]m·xMIIXw·yL·zH2O,其反应如式(1)。
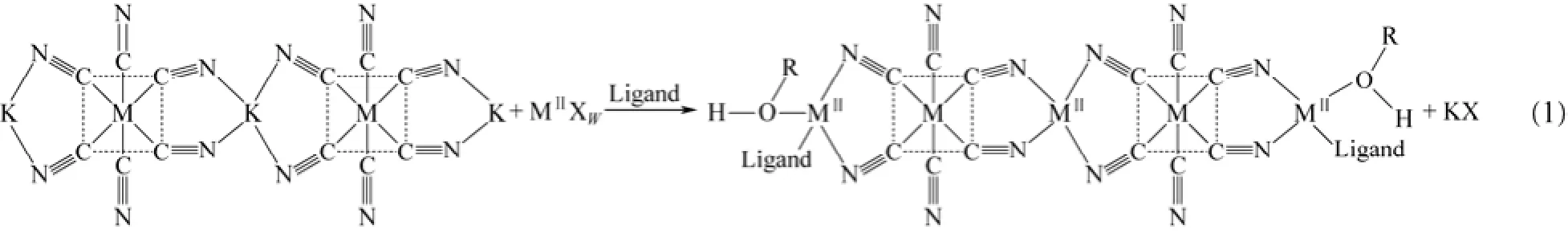
式中,MII为二价金属,可以是 Zn2+、Fe2+、Ni2+、Co2+等;M是过渡金属离子,如Co2+、Co3+、Fe2+、Fe3+、Cr3+、Ni2+等;MIIXw为水溶性金属盐;L为有机配体,可以是醇、醛、酮、醚、酯、酰胺、腈、硫化物及其混合物。
MII与M通过氰基桥连形成三维网络结构,这种天然的网络结构在气体分离、储氢、催化等领域应用广泛。当量的金属盐与金属氰酸盐反应得到的产物对环氧化合物均聚及共聚反应均没有效果;过量的金属盐与金属氰酸盐反应得到的产物 M金属位的配位是不饱和的,使其对环氧烷烃聚合反应具有一定的活性。DMC催化剂的三维网络结构及氰基的强配位效应使其难溶于大多数溶剂,在催化反应中表现为多相催化特征。
目前催化环氧烷烃开环共聚的催化剂按活性金属的不同可分为三类:锌钴系催化剂、锌铁系催化剂以及锌铬系催化剂。其催化环氧烷烃与CO2的共聚机理可分为两步,如图1所示:一是CO2在催化剂活性位上吸附活化,然后环氧烷烃再插入CO2与催化剂的中间;继而再重复该过程,实现链的增长,链的“回咬”形成环状碳酸酯。

图1 环氧烷烃与CO2共聚反应机理
如图2热力学研究表明,形成环状碳酸酯的吉布斯自由能更低,从热力学角度来说,形成环状碳酸酯更加容易进行。同时环氧烷烃与CO2的热力学稳定性也存在差异,DMC催化剂对于环氧烷烃的开环聚合活性比较高。可见选择高活性的催化剂来催化共聚反应显得非常必要,因而对催化剂进行改性,使其能够高效活化CO2,已成为目前研究的热点。
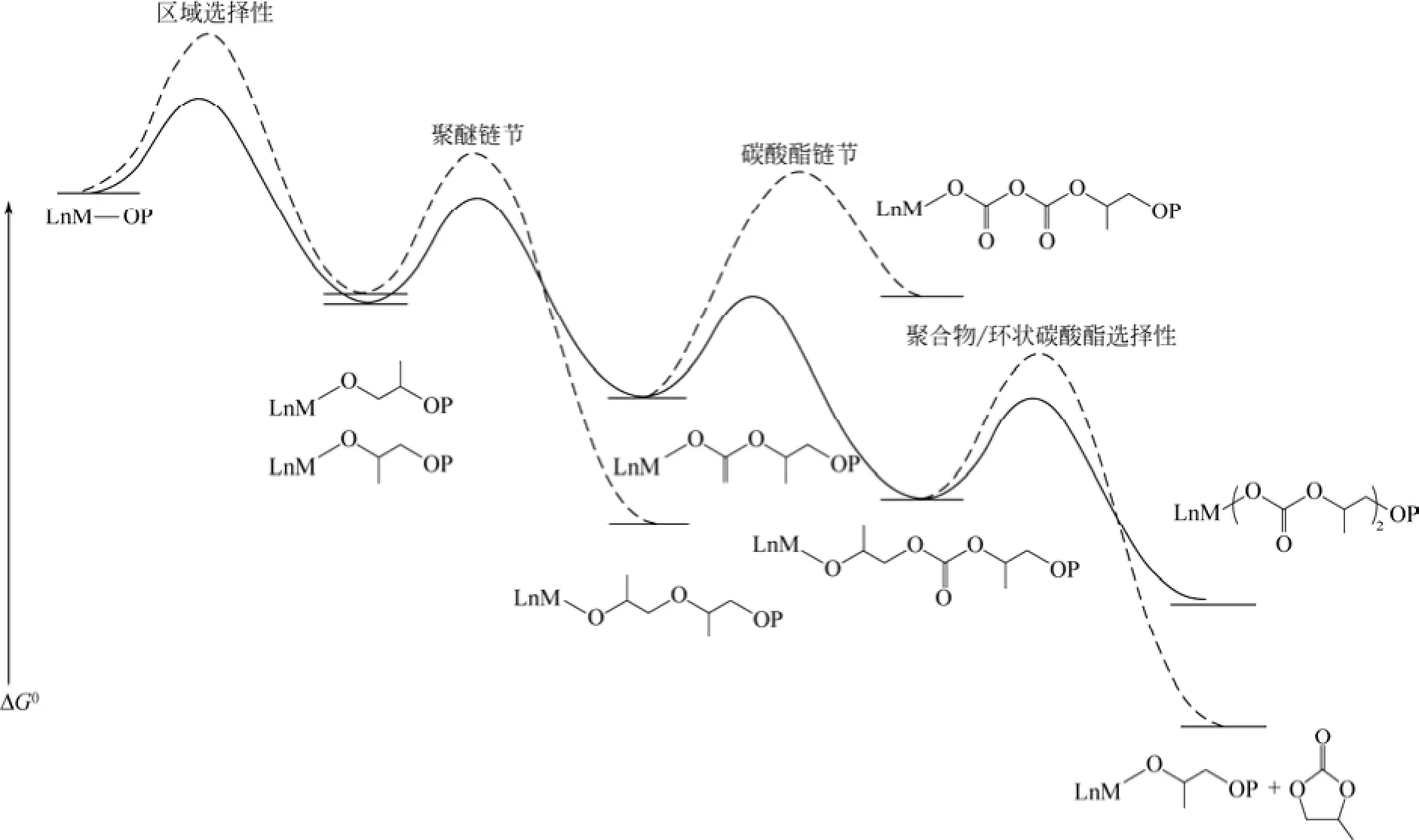
图2 环氧烷烃与CO2共聚反应热力学
1.1 锌钴系双金属氰化络合物催化剂
锌钴系催化剂是最早被合成出来并用于环氧烷烃开环反应的催化剂,因其制备工艺简单、催化效率高、催化剂对空气敏感度不高而在合成聚醚化合物中得到广泛应用。1985年KRUPER等[26]首先将该催化剂引入到环氧烷烃和CO2共聚反应中。
1.1.1 CO2与环氧丙烷共聚
DMC催化剂对PO、环氧环己烷(CHO)与CO2共聚均有很好的催化效果,由于PO与CO2研究较多,在此以PO与CO2共聚为例,其反应如图3所示。

图3 CO2与环氧丙烷共聚
陈上等[27]探索了DMC催化剂催化PO与CO2共聚的工艺条件,发现低温、低用量的催化剂有利于CO2的固定,催化剂的配体不同会影响催化剂效率,但不会改变碳酸酯单元量(FCO2),在最佳工艺条件下,FCO2可以达到31%。然后,他们又研究了采用不溶于水的锌盐来制备催化剂,使得催化效率提高到溶液沉淀法制备的催化剂的两倍[28]。张兴宏等[29]制备出具有纳米层状结构的 DMC催化剂,并将该催化剂用于PO 与CO2的共聚反应,在较低的温度条件下,获得的聚合物相对分子质量可达36500,FCO2可达74.2%,催化效率可达6050g PPC/g Zn。此外,他们通过共聚表观动力学研究了微量水对共聚反应的影响,发现水的存在会与活性锌位配合,从而降低催化剂的活性,导致聚合反应中止,形成更多的环状碳酸酯。并通过实验证明了链引发、链增长和链转移的机理。近年,张兴宏等[30]通过调节烷基的长度可以使获得的聚合物的Tg在–38~6℃可控,通过改变环氧取代基的空间位阻可以使得聚合物的 Tg在 6~84℃可控,并尝试引入可生物降解的纳米纤维素来改善聚合物的性质[31],使得共聚化合物的应用成为可能。相关催化效果如表1所示。
DONG课题组[32]尝试在DMC催化剂的基础上引入稀土三元复合催化剂,两种金属催化剂的协同作用使得FCO2提高到97.1%,聚合物相对分子质量达到10000,Tg随着FCO2的变化在6.7~36.3℃变化,热稳定性也有所提高,原因在于两种催化剂的协同效应使得双金属氰化络合在稀土三元复合催化剂上被活化。高永刚等[33-34]通过加入二羧酸作为链转移剂,并通过调节反应条件来控制FCO2,实现了FCO2在40%~75%变化。通过加入不同相对分子质量的聚丙二醇作为链转移剂可控制相对分子质量在1800~6400变化,FCO2在15.3%~62.5%变化。李志峰等[35]系统研究了复合稀土催化剂组成对共聚反应的影响,发现Y(CCl3COO)3稀土金属配合物有利于共聚反应的发生,当n(Y)/n(Zn) = 6,t=4h时,其催化活性比单纯的DMC催化剂提高了31.5%,聚合物的相对分子质量则没有太大变化。并且相较于单独使用稀土配合物,副产物碳酸丙烯酯的量下降 8%以上,证明了稀土配合物对于活化DMC催化剂的作用。通过添加均苯三酸作为链转移剂,LIU等[36]证明了低聚碳酸酯的结构,并实现了相对分子质量在 1400~3800的可控以及FCO2在20%~54%之间可控。

表1 Zn-Co催化剂催化PO-CO2的聚合效果

表2 Zn-Co改性催化剂催化PO-CO2的聚合效果
SEBASTIAN课题组[37]在制备催化剂的过程中不加入共聚单元,使得催化剂形貌呈现单斜/菱形晶体结构并带有强Lewis酸性,将其应用于PO、CHO和CO2的共聚反应,发现共聚反应没有引发过程,并且活性更高,FCO2可达75%,相对分子质量可达22700,Tg为55℃。这开辟了DMC催化剂应用的新途径。
KIM 课题组[38]系统比较了不同阴离子锌盐作为锌源,叔丁醇和聚(四亚甲基醚二醇)作为配体制备的催化剂的活性,并通过不同表征手段来研究催化剂的结构,将其应用于不同的环氧烷烃(CHO,氧化环戊烯,PO)与CO2共聚反应中,发现所得聚合物相对分子质量适中,相对分子质量分布较窄;同时,该课题组[39]还通过相关表征手段证明了催化剂的结构,即在没有叔丁醇配体时,锌离子因电荷平衡使得其只填补了三分子一个钴氰根离子骨架的空缺,水分子就会进入空腔,配体叔丁醇的存在使得它能够与锌暴露的位点配位,使骨架坍塌,而实验也证明晶体结构的催化剂不能活化PO。
LIU等[40]将有机配体在催化剂制备反应过程中加入反应物中,获得的无定形分散的催化剂的活性更高。VARGHESE等[41]采用H3Co(CN)6作为钴源,使得DMC催化剂的结构方式变化,同时也避免了传统钴盐碱金属盐破坏催化剂的问题,使用1,10-癸二醇作为链转移剂得到的聚合物相对分子质量可达40000,FCO2可达 60%,并实现了相对分子质量在2000~40000可控。陈立班制备出高结晶度的DMC催化剂并将催化剂负载在自制的PBM聚合物上,发现催化效率达 7000g PPC/g 催化剂,FCO2大于40%,相对分子质量大于 30000[42]。相关改性锌钴系催化剂的催化效果如表2所示。
1.1.2 CO2与环氧环己烷共聚
陈上等[43]使用氯化锌作为锌源与钴氰化钾进行反应,叔丁醇作为配体,在90℃、3.8MPa下得到FCO2在40%以上,TOF值达1650h–1的聚碳酸酯。通过动力学研究发现[44],共聚反应对催化剂浓度是一级反应,反应的平均活化能为 41.6kJ/mol。孙学科等[45]通过添加二氧化硅制备出具有纳米层状结构的催化剂,将TOF值提高到3850h–1,相对分子质量达10000,FCO2达44%~47%。通过添加马来酸酐作为第三共聚单体[46],四氢呋喃作为溶剂,将共聚物的相对分子质量提高到 14100,TOF达12700g PPC/g Zn,并实现零副反应。采用羰基硫化物代替CO2与环氧环己烷(CHO)反应,发现无论相对分子质量还是催化效率都不及CO2的活性高[47]。
YI等[48]制备出纳米尺寸的DMC催化剂以及多金属氰化物催化剂,并在催化剂制备过程中添加中性表面活性剂作为乳化剂和配位剂,使得制得的催化剂具有高活性,通过优化反应条件,TOF达41.6g/mol Zn,FCO2达57.8%。利用微波辐射加热方式可使传统的DMC催化剂在0.97MPa下,2~30min内获得相对分子质量达19300,FCO2达75%的聚合物[49]。在传统DMC催化剂基础上,KIM课题组[50]又通过添加其他共配体的方式来制备催化剂,使得制备的催化剂的尺寸在 183~294.2nm可调,催化剂尺寸的差异,使得FCO2在14%~63%变化,相对分子质量在5400~11600变化。
KO等[51]比较了不同反应条件对 CO2/CHO共聚反应的影响,发现增加反应时间、压力、温度以及催化剂的用量都会提高催化活性、聚合物的相对分子质量以及玻璃化转化温度。SRINIVAS课题组[52]系统地比较了采用不同制备方法制备的催化剂活性,并揭示了催化剂结构对反应的影响,发现高密度、强Lewis酸位、合适的结晶度、氯离子以及叔丁醇配体均有利于共聚反应发生。锌-钴催化 CO2与CHO效果如表3所示。
1.1.3 其他
华正江等[53]使用DMC催化剂催化马来酸酐与PO共聚,在较低的催化剂浓度下TOF就可以达到10000g PPC/g 催化剂。同时他们也研究了溶剂对该反应的影响,发现四氢呋喃对于共聚反应的效果最好。然后,他们又研究了邻苯二甲酸酐与PO的共聚反应,发现四氢呋喃作溶剂,90℃下反应3h,转化率就达94%,共聚速度比均聚还快,但相对分子质量不高,共聚速度与单体浓度呈一级关系[54]。使用环氧氯丙烷与CO2进行共聚,FCO2可达70.7%,Tg为31.2℃,环状碳酸酯量较低,热稳定性较好[55]。使用外消旋的氧化苯乙烯作为共聚单体可实现CO2与单体交替聚合,FCO2接近 100%[56]。使用甲基丙烯酸缩水甘油酯作为共聚单体,可实现 FCO2在42.2%~68%可调。在共聚反应中引入 4-甲氧基苯酚,不仅可以抑制甲基丙烯酸缩水甘油酯的自由基均聚,而且可以调控相对分子质量。2,2’-偶氮二异丁腈的存在有利于共聚物的固化[57]。添加内酯有利于提升共聚物的力学性能,为其产品化提供了可能[58]。通过DMC催化剂的催化作用可实现带环氧烷烃终止功能和给予电子烷基的环氧烷烃与CO2进行共聚,TOF为2406g PPC/g Zn,环氧烷烃转化率大于99%。添加带有吸电子基团的氧化苯乙烯,这种新的三元共聚物所特有的吸电子和供电子侧链实现了玻璃化温度在3~56℃可调,使其可以成为可降解的弹性体或塑料[59]。
KIM 课题组[60]比较了 CO2与不同环氧烷烃共聚的活性,发现DMC催化剂对于CO2与氧化环己烯的催化活性最好。其他环氧烷烃得到的聚合物相对分子质量分布也都较窄。采用不同酸酐与PO共聚,发现催化剂活性仍比较高,从而扩展了聚酯多元醇的通用性[61]。
DIENES等[62]采用混合溶胶-凝胶法制备二氧化 硅-双金属氰化物复合催化剂,并将之用于氧化苯乙烯与CO2共聚反应,发现在中等酸性条件下制备的复合催化剂无论从活性或稳定性来看都是最好的。并通过电镜解释了其原因,即活性相由n(Zn)/n(Co)=2.1金属薄片晶体组成。
1.2 锌铁系双金属氰化络合物催化剂
SRINIVAS使用K4Fe(CN)6代替K3Co(CN)6与氯化锌制备DMC催化剂,叔丁醇、三嵌段共聚物EO20PO70EO20作为共配位剂,这种方法制备的催化剂酸性更强,对于活化转化环氧烷烃更加有利[63-64]。同时他们也比较了不同温度条件对于催化剂的影响,发现50℃制得的催化剂活性最好[65]。
表3 Zn-Co催化剂催化CHO-CO2的聚合效果
林强等[66]采用无溶剂球磨法制备DMC催化剂,采用不同的锌盐作为锌源,制备的催化剂的结构类似,并且与溶液沉淀法制备的催化剂结构也类似。将该催化剂应用于PO与CO2的共聚反应,发现聚合物的相对分子质量大于90000,FCO2可达90%。通过加入PEG、PPG、PTMEG等共配体进行固相研磨,制得的催化剂对于共聚反应活性中等,对于环化反应活性较高,但制得的共聚物相对分子质量较高,相对分子质量分布较窄[67]。添加十六烷基三甲基溴来控制制得的催化剂的尺寸在25~79nm,对于CO2与PO环化反应转化率在90%,选择性也较高[68]。
DARENSBOURG 等[69]利用不同的锌盐(ZnCl2、ZnI2)与KCpFe(CN)2CO在水溶液中进行反应,KCpFe(CN)2CO与Zn(CH3CN)4(BF4)2反应,HCpFe(CN)2(phosphine)与 Zn(N(SiMe3)2)2反应制备了一系列的Zn-Fe催化剂,并研究了不同配体对催化剂结构的影响,发现制得的催化剂呈规则的单晶结构,用制得的催化剂催化环氧烷烃与CO2反应,所得的共聚物的FCO2在90%左右。
1.3 其他
张兴宏等[70]系统比较了中心金属及配体对 PO 与CO2共聚反应的影响,发现Zn-Ni催化剂活性最好,阴离子 Br-和 N3-活性适中。陈上等[71]发现Zn-Ni催化剂对于PO共聚反应活性要大于CHO,FCO2也呈现相似的规律,对于这一系列的催化剂,配体对其的影响较其他系列的催化剂要小。
林强等[72]采用球磨的方式制备Zn-Ni和Co-Ni催化剂,制备的催化剂尺寸都是纳米级的,使得催化剂的比表面积较大,活性位更易暴露,从而提升了催化活性。Zn-Ni催化剂催化得到的聚合物相对分子质量为10344,FCO2为83.5%。Co-Ni催化剂催化得到的聚合物相对分子质量为 8478,FCO2为73.7%。而 Zn-Cr催化剂催化产物相对分子质量更高,但FCO2较低。说明该催化剂对于活化CO2的活性不高[73]。
2 催化反应机理
由于DMC催化剂的结构原因,关于它催化环氧烷烃与CO2的机理目前还没有达成共识,文献中描述较多的有3种机理。
孙学科等[45]提出,CHO与CO2共聚可分为两步:即CO2先插入和CHO先插入。配位的CHO可通过Zn-OR基团的亲核攻击而被触发开环,从而产生一个带有末端羟基的新的传播链,然后单体再不断插入Zn-OR基团或Zn-OC(O)OR形成共聚物,其反应机理见图4。
张兴宏等[29]论证了水的存在对聚合反应的影响,如图5所示,中性Lewis碱可以置换活性中心生长的聚合物链,使得共聚物带有碳酸酯自由基和醇端基自由基。锌盐中间体可以固定水,来产生端基带有—OH和—OCOOH的聚合物,带有—OCOOH的聚合物可以通过释放一个CO2变为带有-OH的聚合物。这样的效应会使聚合度下降,因而体系中水的含量直接影响了聚合物的相对分子质量。
GAO等[34]探讨了共配体的加入对反应的影响,如图6所示,PPG可以与中心金属Zn配位,从而释放一个Cl-成为活性位前体S,然后自由Cl去使配位的PO开环形成活性位S*,由于PO的配位是速度控制步骤,链转移剂的配位能力过强,会阻碍PO与Zn酸位的配位,这就证明了诱导期存在的机理。活性位形成后,PO或CO2就可以随机插入Zn-O键中形成传播链。由于PPG在活性Zn位上量比较大,这就需要PPG与传播链之间的迅速转换,以形成一个新的增长链和羟基封端的休眠链,使得PPG参与链增长,而羟基封端的终止链可以可逆参加链转移反应。

图4 Zn-Co·DMC催化剂催化CO2/CHO反应机理图
DMC催化剂催化环氧烷烃与CO2的共聚反应的活性高低在于催化剂的结构差异,通过添加不同的配体,例如叔丁醇等会使催化剂的结构由规整向无定形结构转变,而无定形的催化剂结构在催化反应中可以减少催化剂活化的自由能,有利于催化剂与单体的配位,即催化剂可以通过离去一个离子如Cl-来产生一种电位不饱和的结构,形成一个活性前体,从而可以与环氧烷烃的氧配位使其发生开环反应,形成活性位,然后CO2与环氧烷烃再交替进入活性位,形成链的增长,链的“回咬”或羟基的封端效应可以终止链的增长,形成环状碳酸酯或低聚碳酸酯。而羟基的封端效应的强弱直接影响聚合物的相对分子质量大小,离去基团的强弱直接影响了催化剂与环氧化合物的配位难易。由此可以设想,羟基封端效应过强会使碳链的增长终止,影响相对分子质量增长,离去基团太弱会导致活性中间体难以形成,从而导致催化剂无催化活性。
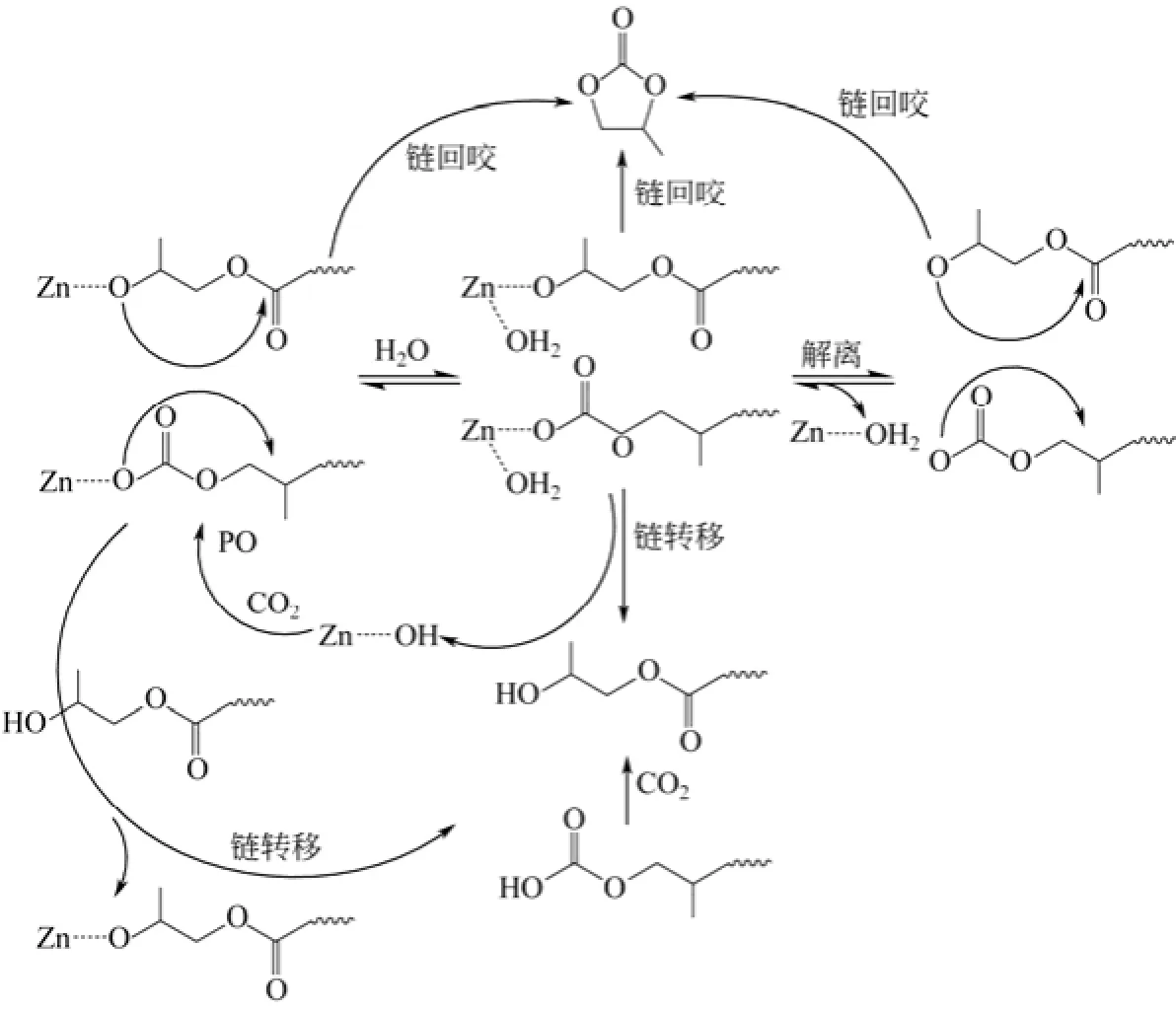
图5 Zn-Co·DMC催化剂催化CO2/PO共聚链转移反应机理
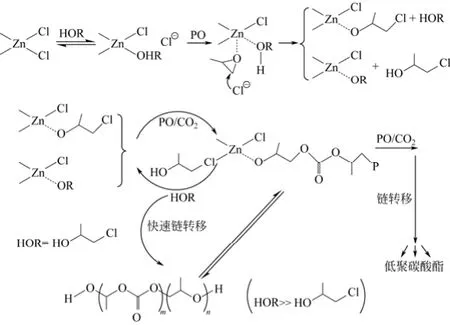
图6 快速链转移反应对链增长的影响机理
3 结论与展望
综上所述,采用不同金属源、原料配比、配体以及不同制备方法都会对催化剂的结构产生重大影响,进而会造成不同催化剂的活性不同。研究发现采用氯化锌和钴氰化钾、叔丁醇为配体溶液沉淀法制备的Zn-Co系催化活性较好。过量的锌源可以使所得的DMC催化剂处于配位不饱和状态,有利于反应物的配位活化。不同的改性方法会对催化效果产生不同的影响,叔丁醇的引入大大提高催化剂的催化效率;稀土的引入可以有效改善PPC中碳酸酯单元的含量,说明稀土的引入可以有效活化CO2,使共聚反应更容易进行;将DMC负载在载体上可以提高聚合物相对分子质量,说明载体的分散作用有利于聚合物相对分子质量以及催化剂活性的提升;对制备方法改进可获得特定晶体结构的催化剂,有利于聚合物相对分子质量的提升;采用不同钴源可以有效避免碱金属对催化剂的毒化作用,从而有效提升碳酸酯单元量,实现对相对分子质量的调控。Zn-Fe系催化剂的研究还不够深入,共聚反应活性不高;其他的催化剂体系都刚刚起步。另外,由于不同的链引发剂和链转移剂在催化剂上的配位能力存在差异,使得采用不同的链引发剂、链转移剂对共聚反应的影响也存在差异,归根结底可以认为活性的差异在于不同方法制备出的催化剂结构存在差异,而催化剂的结构决定了催化活性的高低。
DMC催化剂的结构比较复杂,目前对其精确结构的认识还不充分。从目前的研究来看,过量金属盐与不同配位剂的作用使催化剂产生特殊的表面结构,这是产生催化作用的本质结构原因,其中M处于配位不饱和状态(即含空位)或带有易离去基团,但目前的表征手段还不能原位观察聚合反应时催化剂的表面结构,限制了人们对催化机理的理解。DMC催化剂催化环氧烷烃与CO2共聚反应的速率控制步骤是环氧烷烃的开环,而环氧烷烃的开环反应属于亲核反应,这就使得催化剂配体的选择在催化活性上起到关键的作用,目前含羟基以及醚基的配体在催化聚合反应中活性较好。在聚合反应中,链转移剂的加入可以加快CO2与环氧烷烃的进入催化剂活性结构的速率,使聚合物碳链增长更快。水的存在会对催化剂的端基结构产生破坏,使得CO2的不易被固定,影响催化效率。压力的上升使得CO2的溶解性变强,有利于催化反应,温度的上升可以提供更高的活化能,缩短诱导期的时间,但温度过高会破坏碳酸酯链,影响CO2的嵌入。
再有,以DMC催化剂进行催化反应越来越多,可见 DMC催化剂在催化性能方面有其独特的地方,且对该类催化剂进行改进可以获得满意的结果,具有巨大的应用前景。但是,目前仍有一些问题要解决:①催化剂活性中心结构仍不明确,催化机理还没有形成统一的认识,阻碍了该催化剂的发展;②催化剂的活性和选择性还有待提高;③对于不同的环氧烷烃,催化剂的活性存在巨大差异,相关机理还不明确。因此,揭示DMC催化剂活性中心结构和活性中心环境,乃至解决一些效果不佳的聚合反应,通过对DMC催化剂研究的深入,有望扩大该类催化剂的应用范围,具有潜在的深远意义。
致谢 感谢江苏省“六大人才高峰”高层次选拔培养对象项目资助。
参考文献
[1] GERNGROSS T U,SLATER S C.How green are green plastics?[J].Sci.Am.,2000,283(2):24-29.
[2] ARAKAWA H,ARESTA M,ARMOR J N,et al.Catalysis research of relevance to carbon management: progress,challenges,and opportunities[J].Chem.Rev.,2001,101(4):953-996.
[3] BOLMm C,BECKMANN O,DABARD O A G.The search for new environmentally friendly chemical processes[J].Angew.Chem.Int.Edit.,1999,38(7):907-909.
[4] COOPER A I.Polymer synthesis and processing using supercritical carbon dioxide[J].J.Mater.Chem.,2000,10(2):207-234.
[5] LEITNER W.Homogeneous catalysts for application in supercritical carbon dioxide as a ‘green’ solvent[J].Comptes Rendus de l Academie des Sciences SerieⅡ C:Chimistry,2000,3(7):595-600.
[6] BECKMAN E J.Polymer synthesis-making polymers from carbon dioxide[J].Science,1999,283(5404):946-947.
[7] DARENSBOURG D J,HOLTCAMP M W.Catalysts for the reactions of epoxides and carbon dioxide[J].Coord.Chem.Rev.,1996,153:155-174.
[8] JESSOP P G,IKARIYA T,NOYORI R.Homogeneous catalysis in supercritical fluids[J].Chem.Rev.,1999,99(2):475-493.
[9] KENDALL J L,CANELAS D A,YOUNG J L,et al.Polymerizations in supercritical carbon dioxide[J].Chem.Rev.,1999,99(2):543-563.
[10] NAKAMURA I,INOUE S,ONO T,et al.Chronic arsenical poisoning due to environmental pollution.Seven cases among inhabitants near an abandoned mine[J].The Kumamoto Medical Journal,1976,29(4):172-186.
[11] ROKICKI A,KURAN W.The application of carbon-dioxide as a direct material for polymer synthesis in polymerization and polycondensation reactions[J].J.Macro.Sci.R.M.C.,1981,21(1):135-186.
[12] SUPER M S,BECKMAN E J.Copolymerizations involving carbon dioxide:the use of CO2as a monomer[J].Trends in Polymer Science,1997,5(7):236-240.
[13] WANG D X,KANG M Q,WANG X K.Aliphatic polycarbonates synthesized with carbon dioxide[J].Prog.Chem.,2002,14(6):462-468.
[14] LIU B H,CHEN L B,ZHANG M,et al.Degradation and stabilization of poly(propylene carbonate)[J].Macromol.Rapid.Commun.,2002,23(15):881-884.
[15] Guven O.Radiolysis of a vinyl chloride vinyl acetatecopolymer[J].Angew.Makromol.Chem.,1979,82:217-220.
[16] 李复生,殷金柱,魏东炜,等.聚碳酸酯应用与合成工艺进展[J].化工进展,2002,21(6):395-398.
[17] KAWAGUCHI T,NAKANO M,JUNI K,et al.Examination of biodegradability of poly(ethylene carbonate) and poly(propylene carbonate) in the peritoneal-cavity in rats[J].Chem.& Pharm.Bull.,1983,31(4):1400-1403.
[18] 蒋晶,王小峰,侯建华,等.注射压缩成型聚碳酸酯制品的低温拉伸力学性能[J].化工学报,2015,66(10):4268-4274.
[19] TAKANASHI M,NOMURA Y,YOSHIDA Y,et al.Functional polycarbonate by copolymerization of carbon-dioxide and epoxide-synthesis and hydrolysis[J].Macromol.Chem.Phys.,1982,183(9):2085-2092.
[20] YANG S Y,FANG X G,CHEN L B.Biodegradability of CO2copolymers synthesized by using macromolecule-bimetal catalysts[J].Polym.Adv.Technol.,1996,7(8):605-608.
[21] 陈立班,黄斌.脂肪族聚碳酸酯多醇的制备:1060299[P].1992-04-15.
[22] 王东山,余爱芳,何树杰,等.聚碳酸亚丙酯聚氨酯的合成与性能[J].高分子学报,1999(2):162-166.
[23] 杨淑英,陈立班,余爱芳,等.二氧化碳与环氧化物的调节共聚合反应速率[J].高分子学报,1998(3):83-88.
[24] 方兴高,陈立班,杨淑英.生物降解型聚碳酸顺丁烯二酸亚乙酯1.合成和表征[J].功能材料,1993(5):433-437.
[25] 王景霞,范晓东,周志勇,等.双金属氰化物催化环氧化物开环聚合的研究进展[J].化工进展,2008,27(7):1012-1016.
[26] KUYPER J,BOXHOORN G.Hexacyanometallate salts used as alkene-oxide polymerization catalysts and molecular-sieves[J].J.Catal.,1987,105(1):163-174.
[27] CHEN S,HUA Z J,FANG Z,et al.Copolymerization of carbon dioxide and propylene oxide with highly effective zinc hexacyanocobaltate(III)-based coordination catalyst[J].Polymer,2004,45(19):6519-6524.
[28] CHEN S,ZHANG X H,LIN F,et al.Preparation of double metal cyanide complexes from water-insoluble zinc compounds and their catalytic performance for copolymerization of epoxide and CO2[J].React.Kinet.Catal.Lett.,2007,91(1):69-75.
[29] ZHANG X H,WEI R J,SUN X K,et al.Selective copolymerization of carbon dioxide with propylene oxide catalyzed by a nanolamellar double metal cyanide complex catalyst at low polymerization temperatures[J].Polymer,2011,52(24):5494-5502.
[30] ZHANG X H,WEI R J,ZHANG Y Y,et al.Carbon dioxide/epoxide copolymerization via a nanosized Zinc-Cobalt(Ⅲ) double metal cyanide complex: substituent effects of epoxides on polycarbonate selectivity , regioselectivity and glass transition temperatures[J].Macromolecules,2015,48(3):536-544.
[31] HONG J L,ZHANG X H,WEI R J,et al.Inhibitory effect of hydrogen bonding on thermal decomposition of the nanocrystalline cellulose/poly(propylene carbonate)nanocomposite[J].J.Appl.Polym.Sci.,2014,131(3):1-7.
[32] DONG Y L,WANG X H,ZHAO X J,et al.Facile synthesis of poly(ether carbonate)s via copolymerization of CO2and propylene oxide under combinatorial catalyst of rare earth ternary complex and double metal cyanide complex[J].J.Polym.Sci.Part A:Polym.Chem.,2012,50(2):362-370.
[33] GAO Y G,GU L,QIN Y S,et al.Dicarboxylic acid promoted immortal copolymerization for controllable synthesis of low-molecular weight oligo(carbonate-ether) diols with tunable carbonate unit content[J].J.Polym.Sci.Part A:Polym.Chem.,2012,50(24):5177-5184.
[34] GAO Y G,QIN Y S,ZHAO X J,et al.Selective synthesis of oligo (carbonate-ether) diols from copolymerization of CO2and propylene oxide under zinc-cobalt double metal cyanide complex[J].J.Polym.Res.,2012,19(5):1-9.
[35] 李志峰,秦玉升,赵晓江,等.双金属氰化物/稀土配合物催化二氧化碳和环氧丙烷的共聚合[J].应用化学,2012,29(9):985-989.
[36] LIU S J,QIN Y S,CHEN X S,et al.One-pot controllable synthesis of oligo(carbonate-ether) triol using a Zn-Co-DMC catalyst:the special role of trimesic acid as an initiation-transfer agent[J].Polym.Chem.,2014,5(21):6171-6179.
[37] SEBASTIAN J,DARBHA S.Structure-induced catalytic activity of Co-Zn double-metal cyanide complexes for terpolymerization of propylene oxide,cyclohexene oxide and CO2[J].RSC Adv.,2015,5(24):18196-18203.
[38] KIM I,YI M J,LEE K J,et al.Aliphatic polycarbonate synthesis by copolymerization of carbon dioxide with epoxides over double metal cyanide catalysts prepared by using ZnX2(X = F,Cl,Br,I)[J].Catal.Today,2006,111(3/4):292-296.
[39] BYUN S H,SEO H S,LEE S H,et al.Zn(II)-Co(III)-Fe(III)multi-metal cyanide complexes as highly active catalysts for ring-opening polymerization of propylene oxide[J].Macromol.Res.,2007,15(5):393-395.
[40] LIU H X,WANG X K,GU Y,et al.Preparation and characterization of double metal cyanide complex catalysts[J].Molecules,2003,8 (1):67-73.
[41] VARGHESE J K,PARK D S,JEON J Y,et al.Double metal cyanide catalyst prepared using H3Co(CN)6for high carbonate fraction and molecular weight control in carbon dioxide/propylene oxide copolymerization[J].J.Polym.Sci.Part A:Polym.Chem.,2013,51(22):4811-4818.
[42] 邹志强,张多佑,刘言平,等.高结晶度DMC催化二氧化碳-环氧丙烷共聚[J].高分子材料科学与工程,2010,26(5):5-8.
[43] CHEN S,QI G R,HUA Z J,et al.Double metal cyanide complex based on Zn3Co(CN)6as highly active catalyst for copolymerization of carbon dioxide and cyclohexene oxide[J].J.Polym.Sci.Part A:Polym.Chem.,2004,42(20):5284-5291.
[44] 陈上,张兴宏,戚国荣.Zn-Co双金属氰化络合物催化氧化环己烯/二氧化碳共聚反应[J].催化学报,2006,27(4):355-360.
[45] SUN X K,ZHANG X H,LIU F,et al.Alternating copolymerization of carbon dioxide and cyclohexene oxide catalyzed by silicon dioxide/Zn-CoIIIdouble metal cyanide complex hybrid catalysts with a nanolamellar structure[J].J.Polym.Sci.Part A:Polym.Chem.,2008,46(9):3128-3139.
[46] SUN X K,ZHANG X H,CHEN S,et al.One-pot terpolymerization of CO2,cyclohexene oxide and maleic anhydride using a highly active heterogeneous double metal cyanide complex catalyst[J].Polymer,2010,51(24):5719-5725.
[47] LUO M,ZHANG X H,DU B Y,et al.Alternating copolymerization of carbonyl sulfide and cyclohexene oxide catalyzed by zinc-cobalt double metal cyanide complex[J].Polymer,2014,55(16):3688-3695.
[48] YI M J,BYUN S H,HA C S,et al.Copolymerization of cyclohexeneoxide with carbon dioxide over nano-sized multi-metal cyanide catalysts[J].Solid State Ionics,2004,172(1/2/3/4):139-144.
[49] DHARMAN M M,AHN J Y,LEE M K,et al.Moderate route for the utilization of CO2-microwave induced copolymerization with cyclohexene oxide using highly efficient double metal cyanide complex catalysts based on Zn3Co(CN)6[J].Green Chem.,2008,10 (6):678-684.
[50] LEE I K,HA J Y,CAO C,et al.Effect of complexing agents of double metal cyanide catalyst on the copolymerizations of cyclohexene oxide and carbon dioxide[J].Catal.Today.,2009,148 (3/4):389-397.
[51] OH H J,KO Y S.Effect of polymerization conditions on the polymer properties of CO2-cyclohexene oxide copolymer prepared by double metal cyanide catalyst[J].J.Ind.Eng.Chem.,2013,19(6):1939-1943.
[52] SEBASTIAN J,SRINIVAS D.Effects of method of preparation on catalytic activity of Co-Zn double-metal cyanide catalysts for copolymerization of CO2and epoxide[J].Appl.Catal.A:Gen.,2014,482:300-308.
[53] HUA Z J,QI G R,CHEN S.Ring-opening copolymerization of maleic anhydride with propylene oxide by double-metal cyanide[J].J.Appl.Polym.Sci.,2004,93(4):1788-1792.
[54] 华正江,陈上,方佐,等.双金属氰化络合物催化环氧丙烷和邻苯二甲酸酐共聚[J].高分子学报,2004(4):551-555.
[55] WEI R J,ZHANG X H,DU B Y,et al.Selective production of poly (carbonate-co-ether) over cyclic carbonate for epichlorohydrin and CO2copolymerization via heterogeneous catalysis of Zn-Co (III)double metal cyanide complex[J].Polymer,2013,54(23):6357-6362.
[56] WEI R J,ZHANG X H,DU B Y,et al.Highly regioselective and alternating copolymerization of racemic styrene oxide and carbon dioxide via heterogeneous double metal cyanide complex catalyst[J].Macromolecules,2013,46(9):3693-3697.
[57] WEI R J,ZHANG X H,ZHANG Y Y,et al.Functional poly (carbonate-co-ether) synthesis from glycidyl methacrylate/CO2copolymerization catalyzed by Zn-Co(III) double metal cyanide complex catalyst[J].RSC Adv.,2014,4(7):3188-3194.
[58] LI Y,HONG J L,WEI R J,et al.Highly efficient one-pot/one-step synthesis of multiblock copolymers from three-component polymerization of carbon dioxide , epoxide and lactone[J].Chem.Sci.,2015,6(2):1530-1536.
[59] ZHANG Y Y,WEI R J,ZHANG X H,et al.Efficient solvent-free alternating copolymerization of CO2with 1,2-epoxydodecane and terpolymerization with styrene oxide via heterogeneous catalysis[J].J.Polym.Sci.Pol.Chem.,2015,53(6):737-744.
[60] KIM I,YI M J,BYUN S H,et al.Biodegradable polycarbonate synthesis by copolymerization of carbon dioxide with epoxides using a heterogeneous zinc complex[J].Macromol.Symp.,2005,224:181-191.
[61] SUH H S,HA J Y,YOON J H,et al.Polyester polyol synthesis by alternating copolymerization of propylene oxide with cyclic acid anhydrides by using double metal cyanide catalyst[J].React.& Funct.Polym.,2010,70(5):288-293.
[62] DIENES Y,LEITNER W,MUELLER M G J,et al.Hybrid sol-gel double metal cyanide catalysts for the copolymerisation of styrene oxide and CO2[J].Green Chem.,2012,14(4):1168-1177.
[63] SRIVASTAVA R,SRINIVAS D,RATNASAMY P.Fe-Zn double-metal cyanide complexes as novel,solid transesteritication catalysts[J].J.Catal.,2006,241(1):34-44.
[64] SAIKIA L,SATYARTHI J K,GONNADE R,et al.Double metal cyanides as efficient solid acid catalysts for synthesis of beta-amino alcohols under solvent-free conditions[J].Catal.Lett.,2008,123 (1/2):24-31.
[65] SEBASTIAN J,SRINIVAS D.Novel application of a Fe-Zn double-metal cyanide catalyst in the synthesis of biodegradable,hyperbranched polymers[J].Chem.Commun.,2011,47(37):10449-10451.
[66] ZHANG W Y,LIN Q,CHENG Y,et al.Double metal cyanide complexes synthesized by solvent-free grinding method for copolymerization of CO2and propylene oxide[J].J.Appl.Polym.Sci.,2012,123(2):977-985.
[67] GUO Z F,LIN Q.Coupling reaction of CO2and propylene oxide catalyzed by DMC with co-complexing agents incorporated via ball milling[J].J.Mol.Catal.A:Chem.,2014,390:63-68.
[68] GUO Z F,LIN Q,WANG X H,et al.Rapid synthesis of nanoscale double metal cyanide catalysts by ball milling for the cycloaddition of CO2and propylene oxide[J].Mater.Lett.,2014,124:184-187.
[69] DARENSBOURG D J,ADAMS M J,YARBROUGH J C,et al.Synthesis and structural characterization of double metal cyanides of iron and zinc:catalyst precursors for the copolymerization of carbon dioxide and epoxides[J].Inorg.Chem.,2003,42(24):7809-7818.
[70] ZHANG X H,CHEN S,WU X M,et al.Highly active double metal cyanide complexes:effect of central metal and ligand on reaction of epoxide/CO2[J].Chin.Chem.Lett.,2007,18(7):887-890.
[71] CHEN S,XIAO Z B,MA M Y.Copolymerization of carbon dioxide and epoxides with a novel effective Zn-Ni double-metal cyanide complex[J].J.Appl.Polym.Sci.,2008,107(6):3871-3877.
[72] GUO Z F,LIN Q,ZHU L H,et al.Nanolamellar Zn-Ni/Co-Ni catalysts introduced by ball milling for the copolymerization of CO2with propylene oxide[J].Nanosci.Nanotech.Lett.,2014,6(4):353-356.
[73] LIN Q,GUO Z F,PAN L S,et al.Zn-Cr double metal cyanide catalysts synthesized by ball milling for the copolymerization of CO2/propylene oxide,phthalic anhydride/propylene oxide,and CO2/propylene oxide/phthalic anhydride[J].Catal.Commun.,2015,64:114-118.
第一作者:张鹏(1990—),男,硕士研究生。联系人:刘定华,研究员,主要从事绿色清洁化工新工艺的研究开发工作,侧重于碳酸酯和乙二醇系列产品及催化剂和吸附剂的研究。E-mail ncldh@njtech.edu.cn。
中图分类号:TQ 316.33
文献标志码:A
文章编号:1000-6613(2016)07-2081-10
DOI:10.16085/j.issn.1000-6613.2016.07.020
收稿日期:2015-10-26;修改稿日期:2016-01-14。
The progress of copolymerization of alkylene oxide with carbon dioxide catalyzed by double metal cyanide complex catalysts
ZHANG Peng,LIU Dinghua
(College of Chemistry and Chemical Engineering,Nanjing Tech University,Nanjing 210009,Jiangsu,China)
Abstract:Considering the copolymeration of carbon dioxide (CO2) and alkylene oxides to synthesize aliphatic polycarbonate,we introduced the research progress of double metal cyanide (DMC) complex catalyst used in the process following the comprehensive analysis of the application of aliphatic polycarbonate.In addition,we also analyzed the catalytic effect for different kinds of active center metal combinations in different reaction systems.The Zn-Co-DMC catalyst prepared by precipitation method was used in the catalytic system.By changing the preparation method,ligands and other conditions,we can control the morphology of the catalyst,and thus improve the catalytic activity.The Zn-Fe-DMC catalyst has good activity in the ring-opening of alkylene oxides.At the same time,we also described the combinations of other active center metals for the preparation of DMC catalyst,and got the conclusion that the activity of the copolymerization of CO2and alkylene oxide was strongly affected by the central metal and ligand.Finally,the mechanism of the copolymerization was introduced,and then we pointed out the direction for the design of the DMC catalyst.
Key words:double metal cyanide catalyst;carbon dioxide;reaction mechanism;aliphatic polycarbonate
猜你喜欢
哈哈画报(2022年8期)2022-11-23
中学生数理化·中考版(2022年11期)2022-02-16
中学生数理化·中考版(2021年12期)2021-12-31
中学生数理化·中考版(2021年11期)2021-12-06
今日农业(2021年17期)2021-11-26
小学科学(学生版)(2021年5期)2021-07-22
学生天地(2020年18期)2020-08-25
流程工业(2017年4期)2017-06-21
中学生数理化·中考版(2017年11期)2017-04-18
中学化学(2017年2期)2017-04-01
