
Genetic Diversity and Population Structure for the Conservation of Giant Spiny Frog (Quasipaa spinosa) Using Microsatellite Loci and Mitochondrial DNA
2016-07-25 09:18DannaYURongquanZHENGQinfangLUGuangYANGYaoFUandYunZHANG
Asian Herpetological Research 2016年2期
Danna YU, Rongquan ZHENG,2*, Qinfang LU, Guang YANG, Yao FUand Yun ZHANG
1Institute of Ecology, Zhejiang Normal University, Jinhua 321004, Zhejiang, China2Key Lab of Wildlife Biotechnology and Conservation and Utilization of Zhejiang Province, Jinhua, Zhejiang 321004,China3Jiangsu Key Laboratory for Biodiversity and Biotechnology, College of Life Sciences, Nanjing Normal University,Nanjing 210046, Jiangsu, China
Genetic Diversity and Population Structure for the Conservation of Giant Spiny Frog (Quasipaa spinosa) Using Microsatellite Loci and Mitochondrial DNA
Danna YU1, Rongquan ZHENG1,2*, Qinfang LU1, Guang YANG2,3, Yao FU1and Yun ZHANG1
1Institute of Ecology, Zhejiang Normal University, Jinhua 321004, Zhejiang, China
2Key Lab of Wildlife Biotechnology and Conservation and Utilization of Zhejiang Province, Jinhua, Zhejiang 321004,China
3Jiangsu Key Laboratory for Biodiversity and Biotechnology, College of Life Sciences, Nanjing Normal University,Nanjing 210046, Jiangsu, China
Abstract The giant spiny frog (Quasipaa spinosa) is an endangered species with a relatively small distribution limited to southern China and Northern Vietnam.This species is becoming increasingly threatened because of over-exploitation and habitat degradation.This study provides data on the genetic diversity and population genetic structure of the giant spiny frog to facilitate the further development of effective conservation recommendations for this economically important but threatened species.We examined 10 species-specifc microsatellite loci and Cyt b genes (562 bp) collected from 13 wild populations across the entire range of this species.Results of 10 microsatellite loci analysis showed a generally high level of genetic diversity.Moreover, the genetic differentiation among all 12 populations was moderate to large (overall FST= 0.1057).A total of 51 haplotypes were identifed for Cyt b, which suggests high haplotype nucleotide diversities.Phylogeographic and population structure analyses using both DNA markers suggested that the wild giant spiny frog can be divided into four distinct major clades, i.e., Northern Vietnam, Western China, Central China, and Eastern China.The clades with significant genetic divergence are reproductively isolated, as evidenced by a high number of private alleles and strong incidence of failed amplifcation in microsatellite loci.Our research, coupled with other studies, suggests that Q.spinosa might be a species complex within which no detectable morphological variation has been revealed.The four phylogenetic clades and some subclades with distinct geographical distribution should be regarded as independent management units for conservation purposes.
Keywords Cyt b, Conservation, Giant spiny frog, Microsatellite, Phylogeography, Quasipaa spinosa
1.Introduction
The giant spiny frog (Quasipaa spinosa) is known for the keratinized skin-spines on its chest and the development of hypertrophied forearms as a secondary sex organ in males during the breeding season (Yu et al., 2010).This species inhabits rocky streams in evergreen forests and open fields on mountains 500 m to 1,500 m abovesea level (Zhao, 1998).In Chinese markets, the giant spiny frog is especially valued for its medicinal and nutritional properties, particularly for the high amount of protein contained in its muscles (Ye et al., 1993; Zhao,1998).In addition to harvesting, the habitats of Q.spinosa were destroyed by pollution from upland agriculture and dam and hydropower projects (Chan et al., 2014).Commercial over-exploitation and habitat destruction caused the wild population of this species significantly decline, both in number and distribution (Zhao, 1998).
In 2001, the International Union for the Conservation of Nature (IUCN) Red List registered this species as vulnerable (A2abc) and estimated that over the last threegenerations (approximately 15 years), the wild population had decreased by 30% (Lau et al., 2004; Zhao, 1998).Despite the initiation of domestication and artificial breeding programs in the 1980s, the culture of giant spiny frog remains relatively small in scale because of low fertilization and hatching rates, disease, high overwinter mortality, and inbreeding depression (Chan et al., 2014;Liang et al., 2013).Farming of giant spiny frog mainly depends on the natural supply of tadpoles or adults (Chan et al., 2014; Liang et al., 2013).
Recent phylogenetic study suggested that Q.spinosa may be a cryptic species complex (Che et al., 2009; Ye et al., 2013).However, most frog farms are using frogs from different geographical populations to produce offspring without a detailed record of genetic and performance data (Chan et al., 2014).This practice poses a risk of genetic contamination and a threat of pathogen transmission.Escapees from aquaculture and translocation or even deliberate releases of tadpoles, if not sourced from the same location, have the potential to interbreed with wild frog, thus possibly modifying the genetic structure of wild populations.Interbreeding of Q.spinosa from genetically distinct populations can potentially reduce the fitness and viability of a species,which then becomes increasingly vulnerable to extinction over time (Borrell et al., 2011; Miller et al., 2011).These issues require knowledge of the genetic structure of the species; such knowledge is important for the conservation of indigenous genetic diversity (Chan et al., 2014; Miller et al., 2011).
Information on the intra-specific genetic diversity and relationships among the wild populations of this endangered species remains lacking.Che et al.(2009)suggested that the populations currently under Q.spinosa likely belong to at least three independent evolutionary lineages, but only seven specimens from four localities were used as bases from which to infer phylogenetic relationships.The sample sizes are too small to reflect the genetic diversity and relationships among the populations of Q.spinosa.Ye et al.(2013) also supported that Q.spinosa is a species complex, rather than a single species, on the basis of 12S rRNA and 16S rRNA genes.However, the use of mtDNA alone is insuffcient to refect the quantification and distribution of genetic variation (Dupuis et al., 2012).
In this study, we analyzed data from microsatellite (10 microsatellite loci) and mitochondrial (Cyt b gene) loci collected from Q.spinosa along the entire range of this species to evaluate the phylogeography and population genetics of Q.spinosa, as well as to provide genetic information for further developing effective conservation recommendations to protect this economically important but threatened species.
2.Materials and Methods
2.1 Sample collection and DNA extraction Two hundred and five samples were collected from 13 localities during 2006 and 2007, in China and Vietnam,namely the province (city); Anhui (Huangshan), Fujian (Jianyang, Wuyishan), Hunan (Pingjiang), Guangdong (Yangshan), Guangxi (Longsheng, Yongfu), Jiangxi (Jinggangshan), Yunnan (Pingbian), Zhejiang (Jinhua,Lishui) and Northern Vietnam.Specific spatial details of the collection sites are given in Fig.1 and Table 1.The collection sites were almostly covered the entire geographical range of giant spiny frog (Frost et al., 2006).Some samples were released after obtaining their toe clips.
Muscle or toe clips samples were stored in 95% or 100% ethanol, or frozen at-80°C.The study protocol was reviewed and approved by the Committee of Animal Research Ethics of Zhejiang Normal University.DNA was extracted using the standard phenol-chloroform procedure (Sambrook et al., 1989).

Table 1 Sampling information of giant spiny frog.Geographic sampling localities with their coordinates, number of sample sizes for sample sizes of Cyt b (N) and number of sample sizes for microsatellite (n) are shown.
2.2 Microsatellite genotyping and analysis Among the 15 developed and characterized species-specific genetic markers, 10 proved to be polymorphic and were used in this study (Table S1) (Zheng et al., 2009).Microsatellite genotyping was performed on all individuals from 13 localities, but the Northern Vietnam population could not be included in the analyses because all individuals and loci failed in the amplification.The DNA of Northern Vietnam individuals was extracted several times using a standard phenol-chloroform procedure and DNeasy Tissue Kit (Qiagen), and the electrophoretic band (0.8% agarose) was significantly brighter than 25 ng standard for DNA quantifcation.Amplifcation was performed by gradient temperature polymerase chain reaction (PCR)(44°C to 68°C), but none of the loci can be successfully amplifed.
Amplification was performed in a reaction volume of 25 µL with approximately 0.5 μL genomic DNA,15.88 μL of ddH2O, 2.5 μL of 10× PCR buffer (Mg2+free), 1 μL (10 pmol) of each primer, 2 μL MgCl2(25mM),2 μL dNTP Mixture (2.5mM), and 0.125 μL of TaKaRa Taq (5 U/μL).Following the dye label incorporation procedures from Schuelke (Schuelke, 2000), the forward primer 5′ was appended with a 19 bp M13 sequence (CACGACGTTGTAAAACGAC) and employed at 0.4 μm, whereas the reverse primer used a fluorescentlabeled “M13” primer and employed at 0.4 pmol [either IRD700 or IRD 800(LI-COR)].The PCR thermocycler profile was as follows: 95°C for 5 min followed by 35 cycles at 95 °C for 30 s, and annealing temperature for 30 s (Table S1); 72 °C for 30 s; and a fnal extension step at 72°C for 8 min.The amplifed products were analyzed on a LI-COR4300 automated DNA sequencer using 6.5% denaturing polyacrylamide gels with appropriately labeled 50 bp to 350 bp internal size standard.The gel images were analyzed using the LI-COR SAGA software.
The number of alleles per locus (Na), heterozygosity (H), Allelic frequencies, polymorphic information content (PIC), as well as observed (HO) and expected (HE) heterozygosities were calculated using Cervus 2.0 software (Marshall et al., 1998).Unique alleles, defned as an allele found in only one of the populations, were detected using GDA (Lewis and Zaykin, 2000).The potential deviations from Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were estimated using Arlequin version 2.0 and modified Fisher's exact test (Guo and Thompson, 1992; Schneider et al., 2000).Results of the multiple comparisons were adjusted using Bonferroni correction.
The population genetic differentiation was quantified using the FST-estimator of Weir and Cockerham (Weir and Cockerham, 1984).A Bayesian clustering method was used to organize the dataset and to assign individuals to inferred clusters by using the STRUCTURE software package (Pritchard et al., 2000).Individuals were placed into K clusters using the model-based algorithm, with K chosen in advance.A total of 12 independent runs of K=1 to K=12 were performed at 1,000,000 Markov Chain Monte Carlo (MCMC) repetitions with a 100,000 burn-in period assumed with allele frequencies and combinations without prior information (Du et al., 2012; Pritchard et al., 2000).The log likelihood was used to choose the most likely value for K.The posterior probability K [P(K|X)] was then calculated.Statistic DK, which is the second-order rate of the log probability change of the data between successive values of K, was also estimated (Evanno et al., 2005).The method successfully detected the appropriate number of clusters using multilocus genotype data under a number of gene exchange models.Given that DK could not be evaluated when K = 1, we explored the probability of the K=2 to K=12 on the basis of the above results.
Partitioning genetic diversity through analysis of molecular variance (AMOVA) was performed using ARLEQUIN version 2.0 on the basis of microsatellite data (Schneider et al., 2000).In AMOVA, the total variance was divided into variance among groups, among populations within groups, among individuals within populations, and within individuals.
2.3 Mitochondrial DNA amplification and analysis The Cyt b gene was amplifed with primers CB-10933 (5′-TAT GTT CTA CCA TGA GGA CAA ATA TC-3′) (Simon et al., 1994) and modifed CytbC (5′-CTA CTG GTT GTC CTC CGA TTC ATG T-3′) (Bossuyt and Milinkovitch,2000).Amplifcation was performed in a reaction volume of 50 µL that contained approximately 100 ng of template DNA, 2.5 mM MgCl2, 2.0 mM 10×PCR buffer, 0.4 µM of the forward and reverse primers, 0.2 mM of each dNTP,and 1 U rTaq polymerase (TaKaRa, Dalian, China).The PCR thermocycler profile was as follows: 94 °C for 2 min, followed by 35 cycles at 94 °C for 45 s, 56 °C for 45 s, and 72 °C for 1 min; and a fnal extension step at 72 °C for 10 min.The PCR products were purifed using Wizard PCR Preps DNA purification kit (Promega, Beijing,China).Each fragment was sequenced with an ABI automated DNA sequencer in both directions by using the PCR primers.
All sequences were checked with BioEdit software (North Carolina State University) and by visualinspection.The sequences were then imported into Clustal X (Thompson et al., 1997) for alignment.Genetic diversities for all sampling regions or populations were estimated using haplotype (h) and nucleotide diversities (π), as implemented in DnaSP version 4.0 (Rozas et al.,2003).Sequence divergences between haplotypes and populations were calculated using Arlequin 2.0 (Schneider et al., 2000).Pairwise K2P (Kimura 2-parameter) distances and diagnostic sites for Cyt b gene sequences were calculated in MEGA 5.0 to evaluate the intrapopulation and interpopulation divergence of Q.spinosa (Tamura et al., 2007).The phylogenetic relationship among haplotypes was reconstructed using maximum likelihood (ML) heuristic search methods through by PAUP*4.0 (Swofford, 2002).The robustness of these analyses was assessed using non-parametric bootstrap replications by PAUP*4.0 with 100 replications
ML analyses were performed with the general timereversible (GTR+I+G) model of DNA evolution, which has the shape parameter of a gamma distribution (G)(Posada and Crandall, 1998; Swofford, 2002).The GTR+I+G model was identifed as the best-ftting model under the Akaike information criterion as implemented in Modeltest 3.7 (Posada and Crandall, 1998).In the Bayesian analysis, we used MrBayes 3.1.2 under the GTR+I+G model (Huelsenbeck and Ronquist, 2001).Posterior distributions were determined by MCMC analysis with one cold chain and three heated chains run for 10 million generations with random starting trees.Every 100th tree was sampled.Bayesian analyses were repeated twice, and the same topology was always retrieved.According to the likelihood plots, lnl values stabilized with 10,000 generations.Thus, the frst 100,000 generations were discarded as burn-in.Meanwhile,Bayesian posterior probabilities were calculated according to the remaining set of trees.The rice frog (Fejervarya limnocharis) was chosen as the outgroup for all phylogenetic reconstructions (Che et al., 2009; Liu et al.,2005).
To assess the most probable population confguration and geographical subdivision, hierarchical AMOVA was performed using Arlequin 2.0 (Schneider et al., 2000).The populations were grouped according to different geographical distinctions.Genetic differentiation among samples from different geographic origins was assessed by comparing the average number of pairwise differences between populations (PiXY), the average number of pairwise differences within populations (PiX and PiY), and the corrected average pairwise difference(PiXY_(PiX+PiY)/2).Distances and standard error were calculated using Arlequin 2.0 with 1000 bootstrap replicates (Schneider et al., 2000).
3.Results
3.1 Microsatellite DNA analysis
Genetic variation: All 10 microsatellite loci in the 12 populations showed high genetic polymorphism (Tables S1-S2).However, individuals from Northern Vietnam were either non-functional (not amplifying after repeated attempts) or poorly amplifed (the electrophoretic bands of PCR products were dispersed).A total of 339 different alleles were detected in 205 individuals from 12 populations.The allele sizes at individual loci varied between 12 bp (for locus Psp7) and 92 bp (for locus Psp10).Examination of genotyping errors using MicroChecker revealed no evidence of large allele dropout or stutter-band scoring at any of the 10 loci.The mean number of alleles varied from 6.3 to 15.1.The mean PIC for the 12 populations ranged between 0.63 and 0.88.Within all populations, the HO(0.49-0.72)was lower than HE(0.69-0.91) (Table S2).The 12 populations showed very high gene diversity (mean He=0.82).Among the populations, the gene diversities of XG (He=0.90) and WY (He=0.91) were higher than those of other populations.Deviation from HWE and linkage disequilibrium (LD) was also evaluated in all 12 populations.Out of 120 loci (12 populations multiplied by 10 loci), 65 showed deviation from HWE (P < 0.05;Table S2).All 10 microsatellites were not in LD (P >0.05) for each pair of loci across all samples.
Genetic structure and gene flow: After Bonferroni correction, some multi-loci FSTvalues were small, but all values were significant at P < 0.05 (Table 2).All of the FSTvalues, except for five lower than 0.05, fell between 0.05 and 0.25, indicating that a moderate or large differentiation existed in the populations (Wright, 1978).The average of FSTof all loci was 0.1057.The PJ and HS populations had the largest amount of differentiation at 0.2095.
The clustering method revealed that ∆Kmaxwas 3.02 and peaked at K = 3.The highest posterior probability (ln Pr (X|K) =-4443.6) of the 20 runs for K = 3 was presented along with color-coded probabilities of individuals belonging to a cluster (Figure 1).The highly non-random association of colors showed three distinct genetic cluster groups among the sampling localities.The cluster groups are the Western China, Central China, and Eastern China clusters.The cluster namesrefer to the sample range.The Western China cluster comprised alleles from three populations sampled from Pingbian (PB) of YUNNAN Province, Yangshan (YS) of Guangdong Province, and Longsheng (LH) of Guangxi Zhuang Autonomous Region.The Central China cluster comprised alleles from populations at Yongfu (YF) of Guangxi Zhuang Autonomous Region, Pingjiang (PJ) of Hunan Province, Jinggangshan (JG) and Xingan (XG)of Jiangxi Province, and Wuyishan (WY) of Fujian Province.The eastern China cluster comprised alleles from populations at Jianyang (JY) of Fujian Province,Lishui (LS) and Jinhua (JH) of Zhejiang Province, and Huangshan (HS) of Anhui Province.
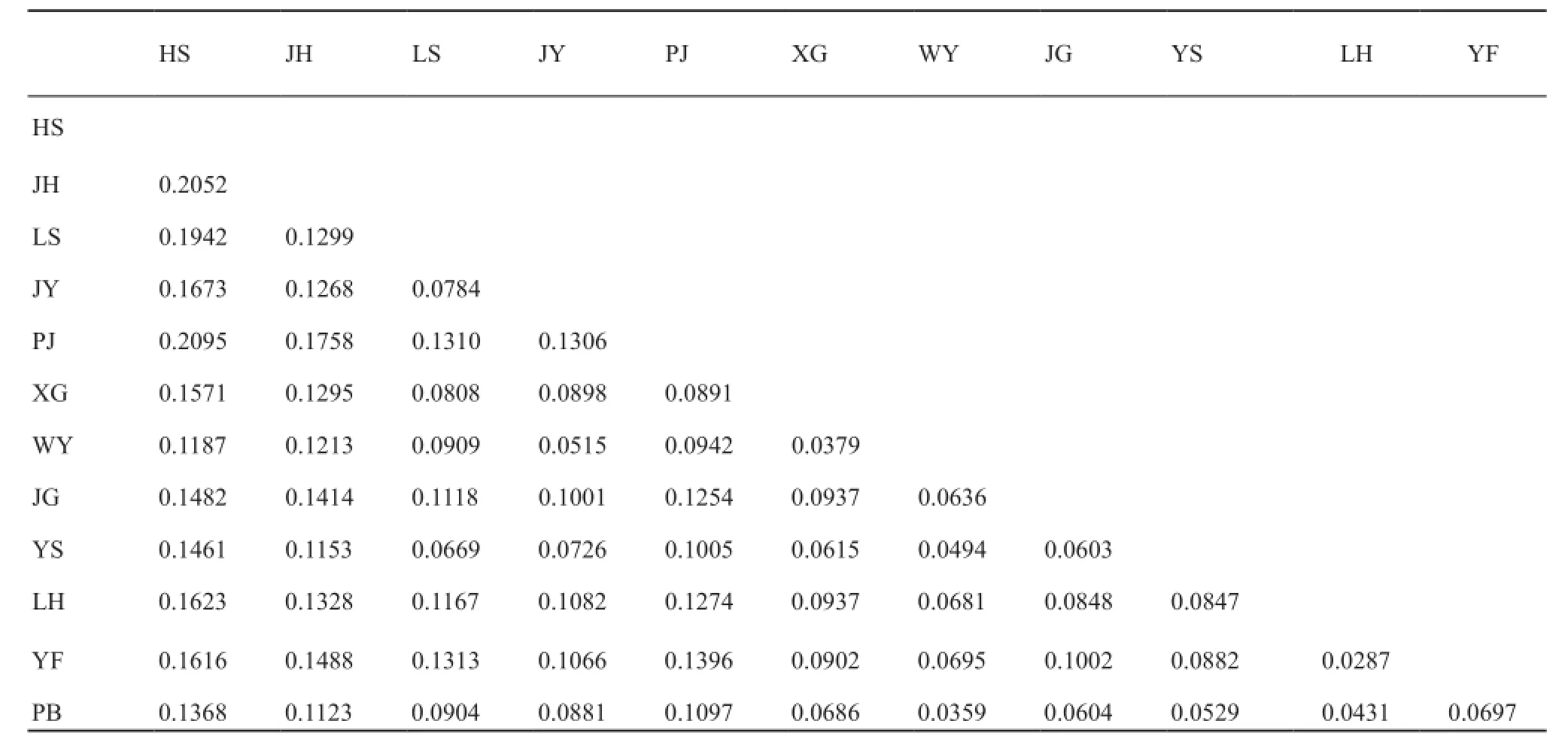
Table 2 Pair-wise FSTbetween the twelve populations estimated from the ten microsatellite loci.
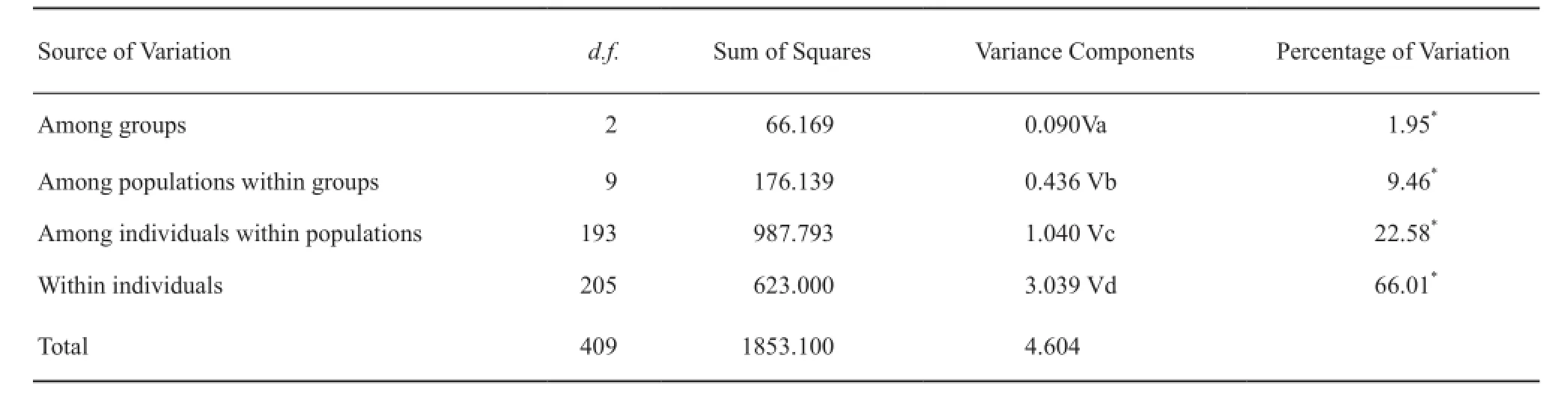
Table 3 Nested Analysis of Molecular Variance (AMOVA) indicating partitioning of genetic diversity.
A total of 118 unique alleles were detected among the three clusters.The Central China cluster possessed the highest number of unique alleles (57.63%), followed by Western China (23.73%) and Eastern China (18.64%)(Table S3).The cluster groups were also analyzed using hierarchical AMOVA method, which revealed the following statistical signifcant (P < 0.001) substructure: 66.01% of the total genetic variations was observed among the individuals, 1.95% among the groups, 9.46% among populations within the groups, and 22.58% among individuals within groups (Table 3).
3.2 Mitochondrial DNA analysis
Sequence variation and genetic distance: A total of 218 variable (38.79%) sites and 211 parsimony-informative (37.54%) sites were found in the 562-bp Cyt b fragments.These sites defined a total of 51 haplotypes (Figure 2).The haplotypic sequences were deposited in GenBank with accession Nos.EU156068-156118.Each population has two to seven haplotypes.Each population also contained unique haplotypes.
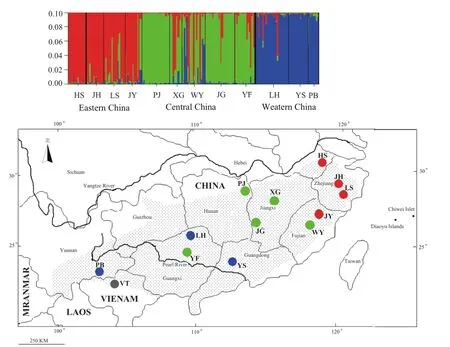
Figure 1 Color scheme: Bayesian cluster analysis of the microsatellite variation among the 12 giant spiny frog populations.Each vertical line represents an individual.The color composition displays the probability of belonging to each of the three clusters as defined by STRUCTURE.The three colors (red, blue, and green) represent the three genetic clusters.Map: Map of sampling localities for this study.The 13 sampled localities have colors corresponding to the genetic cluster wherein the majority of their respective individuals were assigned.Table 1 shows the abbreviation and coordinates of localities.The dotted area shows the distribution of giant spiny frog.
Haplotype and nucleotide diversities were respectively h=0.968±0.006 and π=0.1152±0.0068 for all samples combined.The nucleotide diversity had an overall trend of decline from west to east (Table 4).Estimates of sequence divergence (K2P) among populations ranged from 0.419% (Jianyang, Fujian Province vs.Longsheng,Guangdong Province) to 24.37% (Northern Vietnam vs.Wuyishan, Fujian Province) (Table S4).
Phylogenetic relationship and population structure: The ML and Bayesian inference produced highly similar topologies, which clustered the haplotypes into four major clades, i.e., Northern Vietnam, Western China, Central China, and Eastern China clades (Figure 2).The clade names refer to their geographical pattern.The Northern Vietnam clade has three unique haplotypes, which formed the most basal branch in all phylogenetic reconstructions.Haplotypes H39 and H41 were shared in the Central and Eastern China clades despite having a closer relationship with other haplotypes of the Eastern China clade.Genetic distances among four major clades (range: 7.121-20.027)were almost one order of magnitude higher than those within each group (range: 0.925-8.671; Table 4).To provide a statistical evaluation of the geographical patterns suggested by the phylogenies, all possible sets of AMOVA comparisons were calculated.In the schemes dividing the giant spiny frog populations into two, three,four, and fve groups, the grouping scheme of four groups had a high ΦCTvalue (ΦCT= 0.0715, P < 0.01; Table S5).Genetic distances among the four major clades of giant spiny frogs ranged from 7.12% (Eastern China clade vs.Central China clade) to 20.03% (Northern Vietnam clade vs.Central China clade) (Table 5).
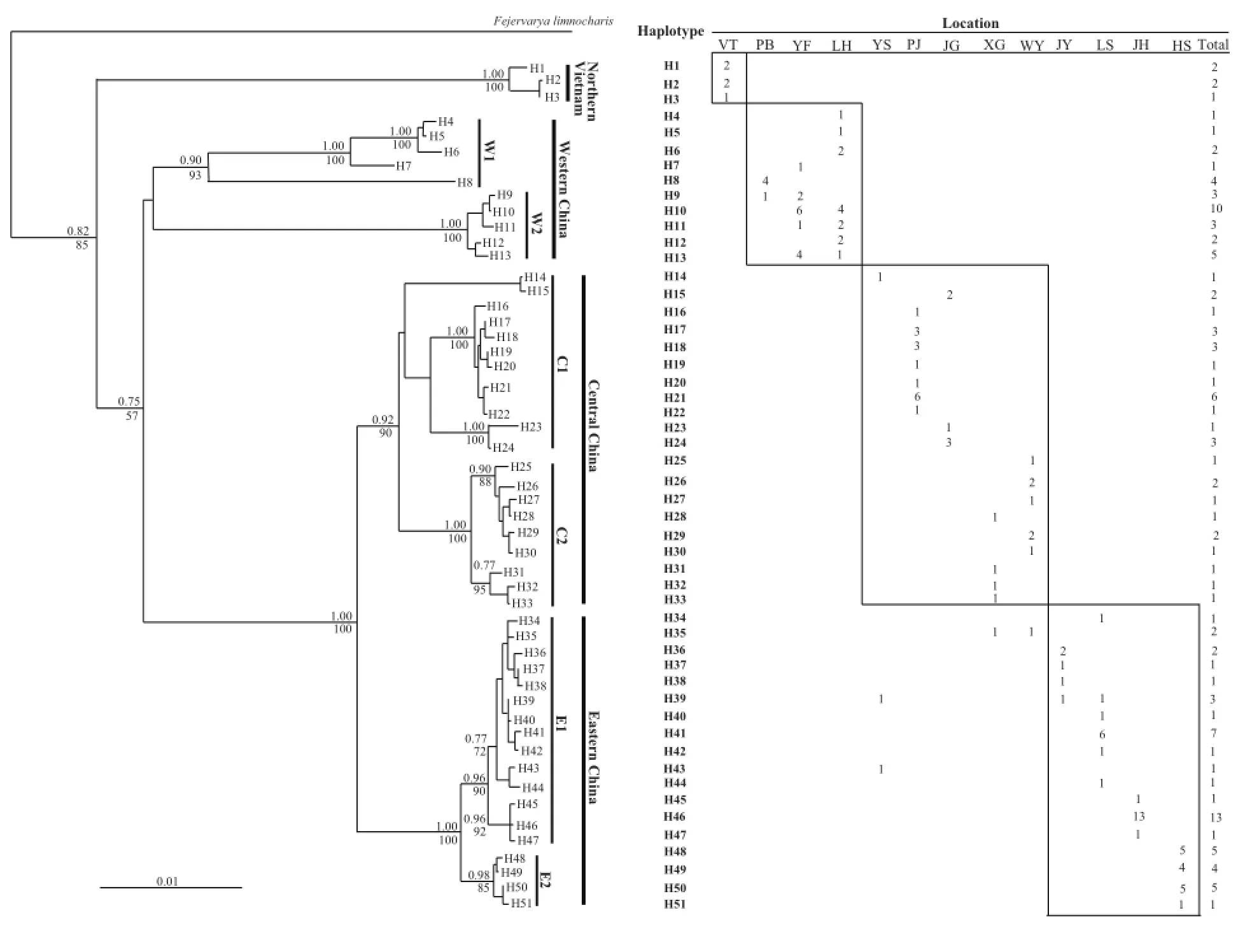
Figure 2 Phylogenetic relationships of haplotypes, and geographic distribution and frequency of each haplotype in the giant spiny frog.Phylogenetic relationships of haplotypes based on the Cyt b gene sequences were derived from Maximum-likelihood (GTR+I+G model)algorithm and Bayesian inference.The numbers above and below the branches are bootstraps and posterior probabilities values, which only shows values higher than 50%.Fejervarya limnocharis was the outgroup species for rooting the tree.
Diagnostic sites: Figure S1 shows the 30 diagnostic sites that clearly distinguished the four major clades.The special base composition at sites 41, 164, 458, and 463 differentiated the alleles H34-51 from the Eastern China clade with all alleles from other three major clades, and the “C” at site 98 only occurred in the haplotypes from the Central China clade (Figure S1).Diagnostic sites were also found between certain subclades, e.g., subclade W1 vs.subclade W2 and subclade E1 vs.subclade E2 (Figure S1).
4.Discussion
4.1 Genetic diversity within Q.spinosa This study is the frst to analyze the genetic diversity and structure of wild giant spiny frog populations using a multiplex set of microsatellite markers.Our result showed that the PIC of the majority of the markers was high (92%, range: 0.23-0.94), which indicated that the 10 microsatellite markers were highly polymorphic and can thus be used for genetic diversity and population structure analyses of giant spiny frog (Botstein et al., 1980).The average allele number of the 10 loci in giant spiny frog was 33.9/locus, which is signifcantly higher than the average allele number in some Anura species (Andersen et al., 2004; Burns et al.,2004).The allele number is usually positively associated with sample size (Goldstein et al., 2005).However, the high polymorphism of the 10 microsatellites enabledthe researchers to use powerful statistical tools to analyze genetic diversity and population structure in 12 populations.HWE is an important prerequisite for population genetic analysis, but the observed allele frequency in more than 50% of the loci in each population was found significantly deviate from HWE in some studies (Musammilu et al., 2014; Zhou et al., 2004).The significant deviation is generally caused by insufficient sample size, null alleles, loss of random mating, and the Wahlund effect (Rousset and Raymond, 1995; Wahlund,1928; You et al., 2008).AMOVA of microsatellites also revealed that variation among populations was 1.59%.Therefore, the Wahlund effect can be excluded because of low genetic differentiation among populations.We detected null alleles in four loci (Psp6, Psp9, Psp10, and Psp14).However, low frequencies (0.08-0.15) of null alleles cannot result in genetic disequilibrium.Nonamplifying samples in repeated trials were not present in any of the primer pairs in the 12 populations of Q.spinosa.The deviation of the loci in this study may be attributed to the relatively small sample size and heterozygote defciencies, which are caused by selection,population mixing, or nonrandom mating (Rousset and Raymond, 1995).

Table 4 The haplotype diversity (h) and nucleotide diversities (π)of Cyt b gene in each population of giant spiny frog.
As shown by the significant departure from HWE at some microsatellite loci, the observed heterozygosities were relatively low in all populations (Table S2).A heterozygote defcit is likely to be apparent in Q.spinosa from the 12 populations, and heterozygosity might be more vulnerable to selection pressures and/or habitat changes (Musammilu et al., 2014).No historical data were available on the genetic diversity and heterozygosity of Q.spinosa but departure of the microsatellite data from HWE expectation in all populations may be attributed to commercial over-exploitation and habitat destruction.Although Q.spinosa is listed as vulnerable by the IUCN and the China Red List (Lau et al., 2004; Zhao, 1998),many populations accessible to hunters may already have been extirpated because such populations may be hunted multiple times per year by multiple hunters (Chan et al.,2014).
Despite using a similar method of analysis, the mean HOlevels found in this study (0.49-0.72) are higher than those found by Andersen et al.(2004) for the Danish European tree frog populations (0.35-0.53) and by Newman and Squire (Newman and Squire, 2001) for the common and widespread Rana sylvatica (0.44-0.50).However, this results of this study are similar to those on the formerly widespread Litoria aurea (0.43-0.82) by Burns et al.(2004).Higher genetic variation in wild giant spiny frog populations can positively affect the protection of giant spiny frog.
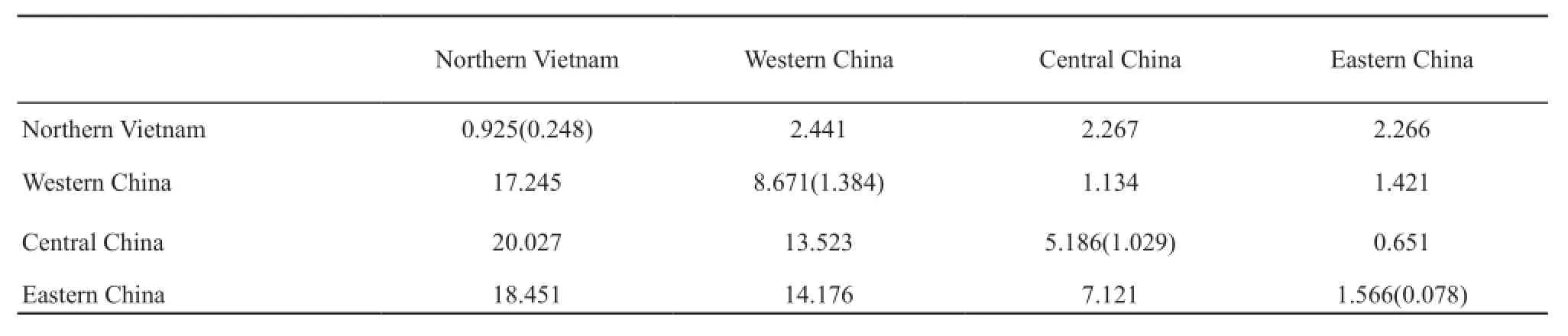
Table 5 Pairwise genetic distances for Cyt b sequences of the giant spiny frog.Below diagonal: Net genetic distances between the four major phylogenetic clades [PiXY-(PiX+PiY)/2].Above diagonal: their standard errors of distances between the four major phylogenetic clades.Values within clades and their standard errors in parenthesis are shown in the diagonal (PiX).
4.2 Phylogeography and population structure Amphibians, especially frogs, have species recognition and mate choice systems that rely on non-morphological characteristics (e.g.advertisement calls) and have the tendency to exhibit conservative morphological evolution.Thus, frogs are considered to have a large amount of cryptic genetic diversity (Bickford et al.,2007; Funk et al., 2012; Stuart et al., 2006).In the Vietnam population, the microsatellite loci were either non-amplifying (Vietnam) or have a high number of private alleles in clades (Table S3).Differentiation at microsatellite loci among clades can indirectly support reproductive isolation.Q.spinosa exhibits high site fdelity, with some individuals found in similar locations in successive years (Chan et al., 2014; Palo et al., 2004).The microsatellite dataset on the 12 giant spiny frog populations (except Vietnam) exhibited a moderate or large genetic differentiation with an overall FSTof 0.1057,which was expected for the frog with low dispersal rates.Q.spinosa is thus expected to exhibit a higher degree of population subdivision than any other major animal taxa because of poor dispersal capabilities (Chan et al., 2014;Palo et al., 2004).STRUCTURE suggested that the frog populations can be split into three distinct genetic clusters,i.e., Western China, Central China, and Eastern China,respectively (Figure 2).Although AMOVA only explained 1.95% of the observed genetic variation among groups,it was statistically significant (P<0.001).The genetic variation confirmed the existence of differentiation among the three groups.The phylogenetic trees using Cyt b gene further corroborated that the four clades are evolutionarily distinct (Figure 2).Relationships among the samples from different geographical locations and putative phylogeographic groups using microsatellite and mitochondrial markers were congruent with the results of Ye et al.(2013).
The clustering result matched the geographic distribution and geological topography of the populations.Giant spiny frogs distributed in the eastern areas are isolated from the frogs in central regions by strong biogeographic barriers, such as Poyang Lake, Gan River,and Wuyi Mountains.The phylogenetic reconstructions based on Cyt b gene also generated a topology that showed a stepwise-ladder pattern (Figure 2).Out of the four clades, the Northern Vietnam clade was the most basal branch, followed by the divergence of Western China clade and the Central and Eastern China clades.The west-to-east dispersal can also be corroborated by the gradual decrease in genetic diversity from west to east.Populations from the original location generally usually lose their genetic diversities during dispersal or expansion (e.g., Yang et al., 2004).
The large number of variable sites (38.79%) and parsimony-informative sites (37.54%) in Cyt b gene also proved that Q.spinosa may be a species complex.Genetic distances among the four major clades of giant spiny frogs are higher than or comparable with those reported in previous studies (Barber, 1999; Bradley Shaffer et al.,2004; Elmer et al., 2007; Yu et al., 2015), as shown by interspecific differentiation in most other frogs.Barber (1999) found mean pairwise distances ranging from 12% to 20% for mitochondrial Cyt b sequences among different Hyla species (i.e., H.arenicolor and H.eximia),which are comparable with the divergences among the four major clades in this study.Some diagnostic sites and unique alleles were also found to distinguish each subclade pair reliably (Figure S1; Table S3).
However, STRUCTURE showed that the YF and YS populations were respectively clustered to the Central and Western China clades.This result differed from those of mitochondrial analysis (Figure 1).Admixture structure also existed in some populations, i.e., YF population mixed with the cluster of Eastern China despite being near the cluster of Western China according to geographical distance.The XG and WY populations also mixed among the three clusters (Figure 1).Discordance between mtDNA and nDNA genes can be attributed to introgression between distinct groups, differences between male and female dispersal rates, or incomplete lineage sorting of ancestral polymorphism (Qu et al.,2012; Yang and Kenagy, 2009).
Our data rejected the possibility that introgression caused the pattern.Introgression always occurred in contact-zone individuals (Yang and Kenagy, 2009).However, admixture was found among far-distance populations, i.e., XG and WY populations showed a mixing among three clusters.Another three populations from Central China only mixed with the cluster of Eastern China.The population of Western China (e.g., LH) mixed with the cluster of Eastern China.As an alternative, we considered that the retention of ancestral polymorphism at the nuclear microsatellite loci likely caused the mixing.The uniparental inheritance of mtDNA resulted in an effective population size four times smaller than that of bi-parentally inherited nuclear DNA.Thus, nuclear DNA is more likely to retain ancestral polymorphism (Yang and Kenagy, 2009).However, further systematic sampling from more areas and more molecular markers should be used in future investigations to confirm the present hypothesis.
4.3 Implications for conservation Our research coupled with other studies suggests that Q.spinosa might be a species complex (Che et al., 2009; Ye et al., 2013).The failure to diagnose biological diversity can hamper conservation efforts and basic scientifc inquiry (Mayden and Wood, 1995).Conservation decisions are often made on the assumption that named taxonomic units represent evolutionary lineages.Phylogeographic analysis provides valuable information on how intraspecific genetic variation is partitioned.Such analysis also identifies evolutionary significant units (ESUs) and management units (MUs) for species.According to the criterion proposed by Moritz (1994), populations that are differentiated for mtDNA haplotypes or at nuclear loci are designated as MUs, whereas those that are reciprocally monophyletic for mtDNA haplotypes and show significant differentiation at nuclear loci are given ESU status.The molecular data presented in this work indicate significant geographical structuring of genetic variation across the range of Q.spinosa, and the topologies of all phylogenetic trees derived from all sequences (Figure 2) showed that reciprocal monophyly criterion for ESU designation was met by the Northern Vietnam, Western China, Central China, and Eastern China clades.
A precise and correct delimitation of species is essential because species are basic units for analysis in biogeography, ecology, macroevolution, biodiversity assessment, and conservation and management (Francesca et al., 2006; Sites Jr and Marshall, 2004).Over- or underresolving species boundaries can result in erroneous decisions for management and conservation (Francesca et al., 2006; Sites Jr and Marshall, 2004).This suggests that a re-evaluation of Q.spinosa boundaries and conservation status is needed.Our results show that Q.spinosa can be divided into multiple ESUs and all of them should be considered as evolutionary independent units, and hence should be independently managed wherever possible to avoid the break-up of coadapted gene complexes and other forms of outbreeding depression (Tallmon et al.,2004; Ficetola and De Bernardi, 2005; Ficetola et al.,2007).At present, most frog farms are using Q.spinosa from different geographical populations, which will lead to a risk of genetic contamination and a threat of pathogen transmission.So, it is necessary to make periodic genetic evaluations of the source population to ensure that they are genetically similar (Borrell et al., 2011).
To protect the relatively abundant genetic variations held in different clades, the four phylogenetic clades and some subclades with distinct geographic distribution may be regarded as independent management units for conservation purposes.
Acknowledgements We are debit to many people who have helped us in collecting samples for this study, in particular Chuntao LIU, Canyang LI, and Xinsheng TANG (Huangshan University).We also want to thank Jiayong ZHANG and Lian CHEN for their assistance in the laboratory work.This research was supported by the National Science Foundation of China (No.31172116 and No.31472015), by the Major Science and Technology Specific Projects of Zhejiang Province of China (No.2010C12008), by the project of the Science Technology Commission of Zhejiang Province of China (No.2011C22006), by the Found for the science and technology innovation team of Zhejiang Province of China (No.2012R10026-07), and by the Key Program of the Twelfth Five Year New Aquatic Varieties Breeding Cooperation of Zhejiang Province of China (2012C12907-9)
References
Andersen L.W., Fog K., Damgaard C.2004.Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea).Proc R Soc Lond B Biol Sci, 271: 1293-1302
Barber P.1999.Phylogeography of the canyon treefrog, Hyla arenicolor (Cope) based on mitochondrial DNA sequence data.Mol Ecol, 8: 547-562
Bickford D., Lohman D.J., Sodhi N.S., Ng P.K., Meier R.,Winker K., Ingram K.K., Das I.2007.Cryptic species as a window on diversity and conservation.Trends Ecol Evol, 22: 148-155
Borrell Y.J., Alvarez J., Blanco G., de Murguía A.M., Lee D.,Fernández C., Martínez C., Cotano U., Álvarez P., Prado J.A.S.2011.A parentage study using microsatellite loci in a pilot project for aquaculture of the European anchovy Engraulis encrasicolus L.Aquaculture, 310: 305-311
Bossuyt F., Milinkovitch M.C.2000.Convergent adaptive radiations in Madagascan and Asian ranid frogs reveal covariation between larval and adult traits.Proc Natl Acad Sci,97: 6585-6590
Botstein D., White R.L., Skolnick M., Davis R.W.1980.Construction of a genetic linkage map in man using restriction fragment length polymorphisms.Am J Hum Genet, 32: 314-331
Bradley S.H., Fellers G.M., Randal V.S., Oliver J.C., Pauly G.B.2004.Species boundaries, phylogeography and conservation genetics of the red-legged frog (Rana aurora/draytonii) complex.Mol Ecol, 13: 2667-2677
Burns E.L., Eldridge M.D., Houlden B.A.2004.Microsatellite variation and population structure in a declining Australian Hylid Litoria aurea.Mol Ecol, 13: 1745-1757
Chan H.K., Shoemaker K.T., Karraker N.E.2014.Demography of Quasipaa frogs in China reveals high vulnerability to widespread harvest pressure.Biol Conserv, 170: 3-9
Che J., Hu J.S., Zhou W.W., Murphy R.W., Papenfuss T.J.,Chen M.Y., Rao D.Q., Li P.P., Zhang Y.P.2009.Phylogeny of the Asian spiny frog tribe Paini (Family Dicroglossidae) sensu Dubois.Mol Phyl Evol, 50: 59-73
Du J., Yan J., Zhou K.2012.Isolation of microsatellite markers for Pelophylax nigromaculata and a tentative application in detecting interspecifc introgression.Gene, 508: 130-134
Dupuis J.R., Roe A.D., Sperling F.A.2012.Multi-locus species delimitation in closely related animals and fungi: one marker is not enough.Mol Ecol, 21: 4422-4436
Elmer K.R., Dávila J.A., Lougheed S.C.2007.Cryptic diversity and deep divergence in an upper Amazonian leaflitter frog,Eleutherodactylus ockendeni.BMC Evol Biol, 7: 247
Evanno G., Regnaut S., Goudet J.2005.Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study.Mol Ecol, 14: 2611-2620
Ficetola G.F., De Bernardi F.2005.Supplementation or in situ conservation? Evidence of local adaptation in the Italian agile frog Rana latastei and consequences for the management of populations.Anim Conserv, 8: 33-40
Ficetola G.F., Garner T., De Bernardi F.2007.Genetic diversity,but not hatching success, is jointly affected by postglacial colonization and isolation in the threatened frog, Rana latastei.Mol Ecol, 16: 1787-1797
Francesca Z., Roberta C., Giuseppe N.2006.Genetic relationships of the western Mediterranean painted frogs based on allozymes and mitochondrial markers: evolutionary and taxonomic inferences (Amphibia, Anura, Discoglossidae).Biol J Linn Soc, 87: 515-536
Frost D.R., Grant T., Faivovich J., Bain R.H., Haas A., Haddad C.F.B., De Sá R.O., Channing A., Wilkinson M., Donnellan S.C., Raxworthy C.J., Campbell J.A., Blotto B.L., Moler P., Drewes R.C., Nussbaum R.A., Lynch J.D., Green D.M.,Wheeler W.C.2006.The amphibian tree of life.Bull Am Mus Nat Hist, 297: 1-370.
Funk W.C., Caminer M., Ron S.R.2012.High levels of cryptic species diversity uncovered in Amazonian frogs.Proc R Soc B,279: 1806-1814
Goldstein P., Wyner Y., Doukakis P., Egan M.G., Amato G.,Rosenbaum H., DeSalle R.2005.Theory and methods for diagnosing species and populations in conservation.Ann Miss Bot Gard, 92: 12-27
Guo S.W., Thompson E.A.1992.Performing the exact test of Hardy-Weinberg proportion for multiple alleles.Biometrics, 48: 361-372
Huelsenbeck J.P., Ronquist F.2001.Mrbayes: Bayesian inference of phylogenetic trees.Bioinformatics, 17: 754-755
Lau M.W.N., Geng B., Gu H., van Dijk P.P., Bain R.2004.Quasipaa spinosa, in: IUCN 2012.IUCN Red List of Threatened Species.Version 2012.1.www.iucnredlist.org
Lewis P.O., Zaykin D.2000.Genetic Data Analysis: computer program for the analysis of allelic data.Version 1.0 (d15).Available at http://hydrodictyon.eeb.uconn.edu/people/plewis/ software.php
Liang Z., Xu Q., Jiang Y., Qin J., Deng W.2013.Situation and development of Quasipaa spinosa in Yongfu County.Guangxi Anim.Husbandry Veterinary Med, 29: 244-246 (in Chinese)
Liu Z.Q., Wang Y.Q., Su B.2005.The mitochondrial genome organization of the rice frog, Fejervarya limnocharis (Amphibia: Anura): a new gene order in the vertebrate mtDNA.Gene, 346: 145-151
Marshall T.C., Slate J., Kruuk L.E.B., Pemberton J.M.1998.Statistical confdence for likelihood-based paternity inference in natural populations.Mol Ecol, 7: 639-655
Mayden R.L., Wood R.M.1995.Systematics species concepts and the evolutionarily significant unit in biodiversity and conservation biology.Am Fisheries Soc Symp, 17: 58-113
Miller P.A., Fitch A.J., Gardner M., Hutson K.S., Mair G.2011.Genetic population structure of Yellowtail Kingfish (Seriola lalandi) in temperate Australasian waters inferred from microsatellite markers and mitochondrial DNA.Aquaculture,319: 328-336
Moritz C.1994.Applications of mitochondrial DNA analysis in conservation: a critical review.Mol Ecol, 3: 401-411
Musammilu K., Abdul-Muneer P., Gopalakrishnan A., Basheer V., Gupta H., Mohindra V., Lal K.K., Ponniah A.2014.Identification and characterization of microsatellite markers for the population genetic structure in endemic red-tailed barb,Gonoproktopterus curmuca.Mol Boil Rep, 41: 3051-3062
Newman R.A., Squire T.2001.Microsatellite variation and fnescale population structure in the wood frog (Rana sylvatica).Mol Ecol, 10: 1087-1100
Palo J.U., Schmeller D.S., Laurila A., Primmer C.R., Kuzmin S.L., Merilä J.2004.High degree of population subdivision in a widespread amphibian.Mol Ecol, 13: 2631-2644
Posada D., Crandall K.A.1998.Modeltest: testing the model of DNA substitution.Bioinformatics, 14: 817-818
Pritchard J.K., Stephens M., Donnelly P.2000.Inference of population structure using multilocus genotype data.Genetics,155: 945-959
Qu Y., Zhang R., Quan Q., Song G., Li S.H., Lei F.2012.Incomplete lineage sorting or secondary admixture: disentangling historical divergence from recent gene fow in the Vinous-throated parrotbill (Paradoxornis webbianus).Mol Ecol,21:6117-6133
Rousset F., Raymond M.1995.Testing heterozygote excess and defciency.Genetics, 140: 1413-1419
Rozas J., Sánchez-DelBarrio J.C., Messeguer X., Rozas R.2003.DnaSP, DNA polymorphism analyses by the coalescent and other methods.Bioinformatics, 19: 2496-2497
Sambrook J., Fritsh E.F., Maniatis T.1989.Molecular Cloning: a Laboratory Manual, second edition.New York, England: Cold Spring Harbor Laboratory Press
Schneider S., Roessli D., Excoffier L.2000.Arlequin Ver.2.001: a Software for Population Genetic Data Analysis.Genetics and Biometry Laboratory, Department of Anthropology, University of Geneva, Switzerland
Schuelke M.2000.An economic method for the fluorescent labeling of PCR fragments.Nat Biotechnol, 18: 233-234
Simon C., Frati F., Beckenbach A., Crespi B., Liu H., Flook P.1994.Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers.Ann Entomol Soc Am, 87: 651-701
Sites Jr J.W., Marshall J.C.2004.Operational criteria for delimiting species.Ann Rev Ecol Syst, 35: 199-227
Stuart B.L., Inger R.F., Voris H.K., 2006.High level of cryptic species diversity revealed by sympatric lineages of Southeast Asian forest frogs.Biol Lett, 2: 470-474
Stuart S.N., Chanson J.S., Cox N.A., Young B.E., Rodrigues A.S., Fischman D.L., Waller R.W.2004.Status and trends of amphibian declines and extinctions worldwide.Science, 306: 1783-1786
Swofford D.L.2002.PAUP*: phylogenetic analysis using parsimony, Version 4.Sinauer Associates, Sunderland,Massachusetts
Tallmon D.A., Luikart G., Waples R.S.2004.The alluring simplicity and complex reality of genetic rescue.Trends Ecol Evol, 19: 489-496
Tamura K., Dudley J., Nei M., Kumar S.2007.MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0.Mol Biol Evol, 24: 1596-1599
Tamura K., Peterson D., Peterson N., Stecher G., Nei M.,Kumar S.2011.MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods.Mol Boil Evol, 28: 2731-2739
Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F.,Higgins D.G.1997.The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools.Nuc Acid Res, 25: 4876-4882
Wahlund S.1928.Zusammensetzung von populationen und korrelationserscheinungen vom standpunkt der vererbungslehre aus betrachtet.Hereditas, 11: 65-106
Weir B.S., Cockerham C.C.1984.Estimating F-statistics for the analysis of population structure.Evolution, 38: 1358-1370
Wright S.1978.Evolution and the genetics of populations.Chicago, America: University of Chicago Press
Xie F., Lau M.W.N., Stuart S.N., Chanson J.S., Cox N.A.,Fischman D.L.2007.Conservation needs of amphibians in China: A review.Sci China C, 50: 265-276
Yang D.S., Kenagy G.J.2009.Nuclear and mitochondrial DNA reveal contrasting evolutionary processes in populations of deer mice (Peromyscus maniculatus).Mol Ecol, 18: 5115-5125
Yang Y.H., Zhang D.X., Li Y.M., Ji Y.J.2004.Mitochondrial DNA diversity and preliminary biogeography inference of the evolutionary history of the black-spotted pond frog Rana nigromaculata populations in China.Acta Zool Sin, 50: 193-201
Ye C., Fei L., Hu S.1993.Rare and economic amphibians of China.Chengdu, China: Sichuan Publishing House of Science and Technology (in Chinese)
Ye S.P., Huang H., Zheng R.Q., Zhang J.Y., Yang G., Xu S.X.2013.Phylogeographic Analyses Strongly Suggest Cryptic Speciation in the Giant Spiny Frog (Dicroglossidae: Paa spinosa) and Interspecies Hybridization in Paa.PloS one,8:e70403
You E.M., Chiu T.S., Liu K.F., Tassanakajon A., Klinbunga S., Triwitayakorn K., de La Peña L.D., Li Y., Yu H.T.2008.Microsatellite and mitochondrial haplotype diversity reveals population differentiation in the tiger shrimp (Penaeus monodon)in the Indo-Pacifc region.Anim Genet, 39: 267-277
Yu B.G., Zheng R.Q., Zhang Y., Liu C.T.2010.Geographic variation in body size and sexual size dimorphism in the giantspiny frog Paa spinosa (David, 1875) (Anura: Ranoidae).J Nat Hist, 44: 1729-1741
Yu D.N., Zhang J.Y., Peng L., Shao C., Zheng R.Q.2015.Do cryptic species exist in Hoplobatrachus rugulosus? An examination using four nuclear genes, the Cyt b Gene and the complete mt genome.PLoS One 10: e0124825
Zhao E.M.1998.China Red Data Book of Endangered Animals-Amphibia.Beijing, China: Science Press (in Chinese).
Zheng R.Q., Ye R.H., Yu Y.Y., Yang G.2009.Fifteen polymorphic microsatellite markers for the giant spiny frog, Paa spinosa.Mol Ecol Resour, 9: 336-338
Zhou J., Wu Q., Wang Z., Ye Y.2004.Genetic variation analysis within and among six varieties of common carp (Cyprinus carpio L.) in China using microsatellite markers.Russ J Genet (Genet), 40: 1144-1148
E-mail: zhengrq@zjnu.cn
Received: 10 November 2015 Accepted: 25 February 2016
DOI:10.16373/j.cnki.ahr.150040
*Corresponding author:Dr.Rongquan ZHENG, from Zhejiang Normal University, with his research focusing on amphibian molecular ecology.
 Asian Herpetological Research2016年2期
Asian Herpetological Research2016年2期
- Asian Herpetological Research的其它文章
- Tracing the Origin of the Black-spotted Frog, Pelophylax nigromaculatus, in the Xinjiang Uyghur Autonomous Region
- Coevolution of Male and Female Response Preferences to Sexual Signals in Music Frogs
- Ecological Niche Divergence between Trapelus ruderatus (Olivier,1807) and T.persicus (Blanford, 1881) (Sauria: Agamidae) in the Middle East
- The Effects of Chronic Hypoxia on Thermoregulation and Metabolism in Phrynocephalus vlangalii
- Comparison of Skull Morphology in Two Species of Genus Liua (Amphibia: Urodela: Hynobiidae), L.shihi and L.tsinpaensis
- Discovery of Female Laudakia papenfussi Zhao, 1998, with Insights into its Phylogenetic Relationships