
叶黄素聚集体吸收光谱的实验分析与理论模拟
2016-07-12 12:55卢礼萍刘桂玲魏良淑
光谱学与光谱分析 2016年10期
卢礼萍,李 明,刘桂玲,魏良淑,吴 芳
南京农业大学理学院,江苏 南京 210095
叶黄素聚集体吸收光谱的实验分析与理论模拟
卢礼萍,李 明,刘桂玲,魏良淑,吴 芳*
南京农业大学理学院,江苏 南京 210095
分子聚集体表现出单分子所不具备的特有功能,利用吸收光谱对聚集体分子结构特性的研究是理解其电子和能量转移功能的基础。实验中利用紫外-可见分光光度计,检测了叶黄素在乙醇溶液中的单体吸收谱和在1∶1乙醇水溶液中的聚集体吸收谱。并通过对叶黄素单体吸收谱的高斯分解,获得了激发态的振动能级结构参数。理论上采用时间相关函数描述的吸收谱线和Frenkel激子模型,通过模拟单分子吸收光谱,获得了叶黄素分子的激发能、特征模振动频率、Huang-Phys因子等参数。再利用这些参数进行了聚集体分子光谱的模拟,分析了叶黄素聚集体的分子结构影响光谱变化的原因。结果表明,(1) 分子间的相互作用是决定吸收光谱峰位移动的主要原因,实验中叶黄素H-聚集体的吸收光谱较单体蓝移了77 nm,模拟显示相互作用在2 000 cm-1附近;(2) 聚集体分子个数越多,聚集协作效应越大,吸收光谱半高宽变小,同时吸收峰进一步蓝移;(3) 环境的无序度对吸收光谱的半高宽也存在较大的影响,无序度越大,吸收光谱半高宽越大。实验结果为进一步研究聚集体在生物系统和材料系统中的功能提供了理论依据。
H-聚集体;Frenkel激子;吸收光谱;协作效应
引 言
在高等植物和大部分藻类的光合作用中,类胡萝卜素分子扮演着辅助捕获光能和保护光合器官两大角色[1]。在动物和人体中,它们通过淬灭单线态氧来捕获自由基,具有抗氧化作用[2-3]。叶黄素是分子两端含紫罗兰酮环的二羟基类胡萝卜素。大量流行病学证据表明,叶黄素在预防癌症及心血管疾病等方面起着重要的作用,是目前功能性食品和药品成分研究的一个热点[4]。然而生物功能器官中存在着大量的水,在含水环境中叶黄素的存在状态和物理化学性质受到了越来越多的关注。研究表明,叶黄素分子在含水的有机溶剂中或在双层脂膜中形成聚集体[5],这种聚集体的物理和化学性质对其生物功能有着重要的影响。
目前对单体类胡萝卜素分子吸收光谱的理论和实验研究比较成熟[6]。普遍认为单体的类胡萝卜素分子具有两个激发态S1和S2,由于对称性,只有基态S0到S2的跃迁是允许的[7]。研究表明,类胡萝卜素分子聚集后,形成的H-聚集体吸收光谱蓝移,J-聚集体吸收光谱红移[8]。无论是哪种聚集,分子间存在较强的相互作用,通常采用Frenkel激子来模拟分子间的强相互作用。深入理解聚集体光谱性质的变化,则需要将聚集体光谱与相关理论结合考察,目前此项研究尚不充分。该研究采用理论模拟实验的方法,分析了影响聚集体分子吸收光谱的因素,对深入研究类胡萝卜素分子在生物器官中发挥的生物学功能提供了参考。
1 实验部分
UV 1700紫外-可见分光光度计(SHIMADZU UV1700,日本岛津公司),扫描范围350~550 nm。叶黄素(92%)购买于上海源叶生物科技有限公司,溶解于乙醇中形成浓度为50 μmol·L-1的叶黄素单体溶液。分别取两份5 mL该单体溶液,第一份加入5 mL去离子水,在1∶1的乙醇水溶液中形成聚集体;第二份加入5 mL乙醇,形成等浓度的单体溶液。用紫外-可见分光光度计测定各样品的吸收光谱。
2 理论部分
2.1 分子吸收光谱
由量子力学可知,初态|k〉到末态|l〉的跃迁速率由Fermi黄金定则决定
(1)
根据线性吸收光谱理论和涨落耗散定理[9],吸收光谱的波形可用跃迁偶极矩的关联函数表示,环境的作用采用谐振子模型来处理,考虑到振动能级,吸收光谱波形表达式[10]。
(2)
μ(t)是Heisenberg表象中的偶极距。其中
(3)
式(3)第一项考虑到了光谱线的展宽效应,第二项为分子振动的影响,λ是分子与环境的耦合强度,Λ为环境涨落衰减速率,wi为特征模振动频率,si是电子-振动耦合强度Huang-Phys因子。
2.2 聚集体分子吸收光谱
采用Frenkel激子模型研究聚集体系统,设每个分子都是二能级系统,聚集系统的哈密顿量为[11]
(4)
|n〉表示第n个分子被激发,其他分子处于基态。Ei表示第i个单分子激发能。Di表示系统由于受到环境的涨落影响产生的无序度,取值满足均值为0,标准偏差为Di的高斯分布,取值之间无关联。Vnm是第n和第m个分子之间的偶极相互作用,设V不随环境涨落,可以表示为式(5)
(5)
对式(4)哈密顿量对角化,可得到各激发态能量和波函数式(6)。
(6)
聚集体的跃迁距和局域化强度为式(7)
(7)
聚集体振动分辨的吸收光谱波形为式(8)
(8)
式(8)中gνν(t)=ζνg(t)。
3 结果与讨论
3.1 叶黄素单体光谱
实验首先检测了乙醇溶液中的叶黄素单体吸收光谱,并对其进行高斯分解,见图1。图1中叶黄素单体在350~550 nm范围的吸收对应S0→S2的跃迁。高斯分解结果显示,该吸收带存在四个吸收子峰,分别位于393,420,447和476 nm,对应的是S2激发态上各个振动能级0→3,0→2,0→1和0→0的吸收。
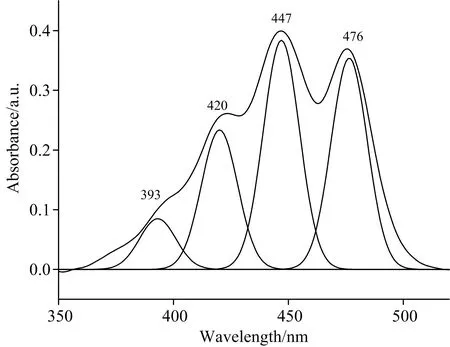
图1 叶黄素吸收谱
由上述分析可知,吸收光谱中存在多个振动带。通过分析图1中最大的高斯峰与其他三个波峰之间的波数差,得到三个振动特征频率w1=1 438 cm-1,w2=1 363 cm-1,w3=3 074 cm-1;通过三个高斯峰值与最大峰值的面积比值[12],确定上述振动特征频率对应的Huang-Rhys因子s1=0.791,s2=0.348,s3=0.123。取第二激发态中心激发能E=22 900 cm-1,λ=700 cm-1,Λ=40 cm-1。利用式(2)模拟了叶黄素单体的吸收谱,见图2(a)。从模拟的结果不难发现,在低能量振动的长波方向,模拟结果与实验值基本一致;在高能量振动的短波方向,可能还存在多个微扰振动的连续吸收带,理论与实验存在一定的差别。为了研究环境对吸收谱产生的影响,模拟了不同无序度D时的吸收谱,见图2(b)。图2(b)表明,无序度越大,该S2电子能级上的最大振动带越宽,有可能延伸至其他振动带,从而把其他振动带掩盖,吸收光谱中其他振动带的吸收不明显。
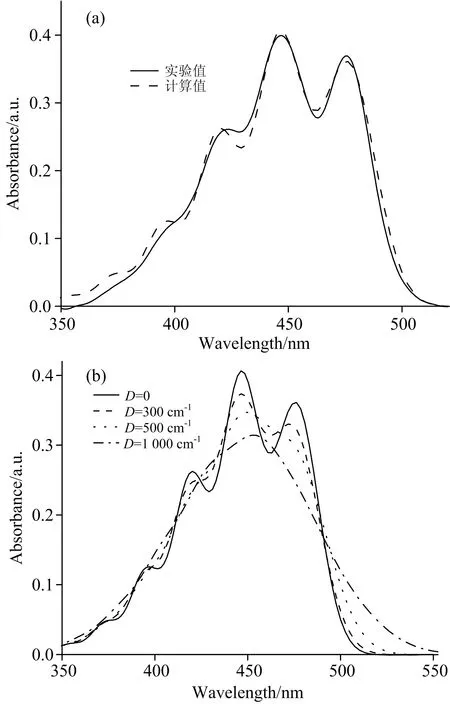
图2D=0时,叶黄素单体吸收谱的理论计算和实验值比较(a);不同无序度时叶黄素单体吸收谱的理论计算(b)
Fig.2 Comparison of theoretical and experimental absorption spectra of lutein monomer withD=0 (a);Theoretical absorption spectra of lutein monomer with differentD=300 cm-1(b)
3.2 叶黄素H-聚集体的吸收光谱
在1∶1体积的乙醇-水溶液中,叶黄素形成聚集体,其吸收光谱见图3。图3显示,在聚集体形式下叶黄素吸收光谱从单体最大吸收447 nm蓝移至370 nm,产生了77 nm的蓝移,同时损失了振动能量,峰形变窄,半高宽度为37 nm。可见在该溶液体系中,叶黄素分子以平行的方式紧密排列,形成了H-聚集体。根据文献报道,分子间距大约为0.5 nm[13]。
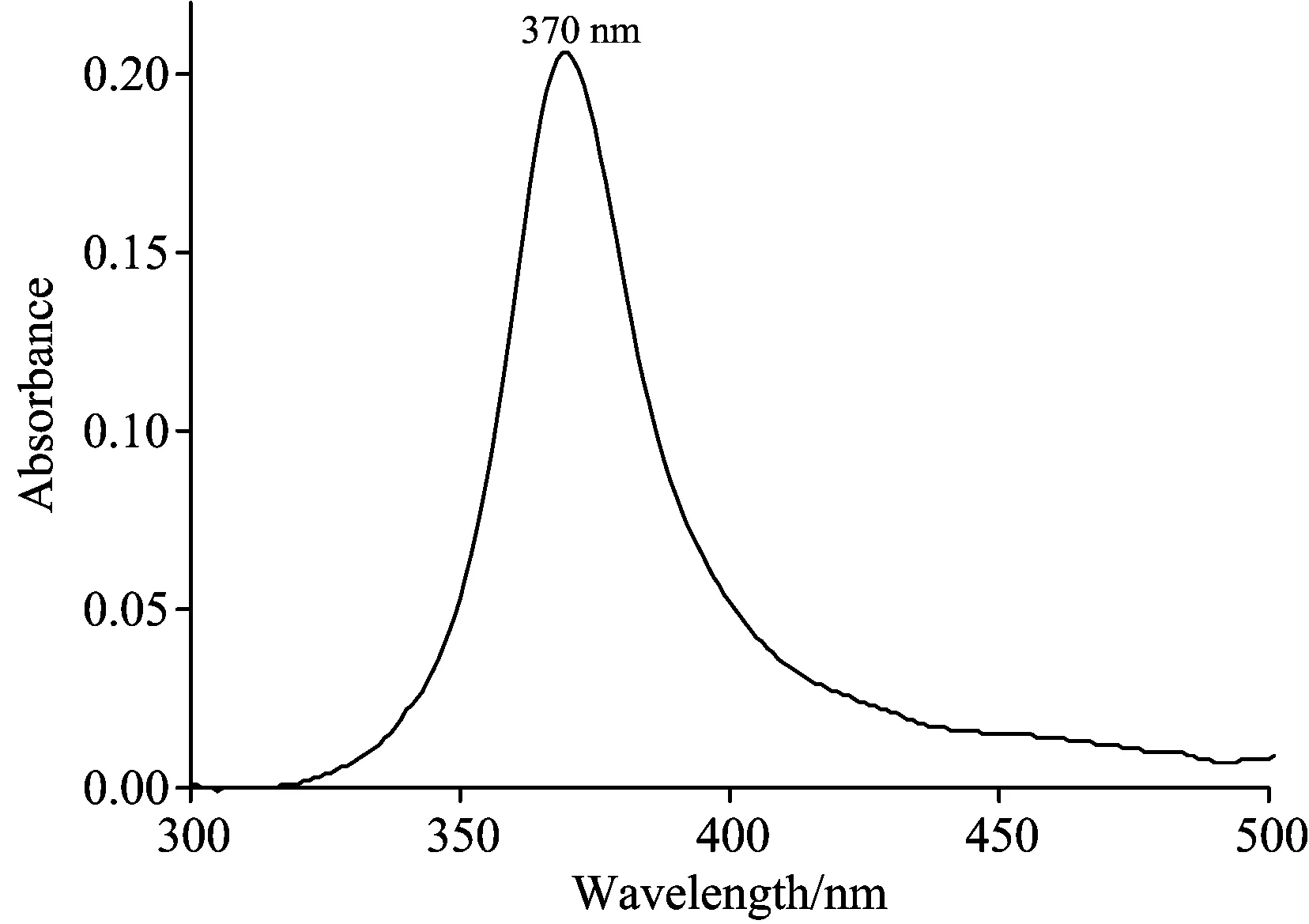
图3 叶黄素H-聚集体的吸收谱的实验值
利用分子的吸收光谱,可判断分子聚集体的形式。要获得具体的聚集体结构信息,则要更精确的实验和可靠的理论。叶黄素聚集体中,分子以平行方式排列。利用式(5)即可得到分子间的偶极相互作用,取r=0.5nm,μ=5 Debye。在0~π区间内,β小于32°时,偶极相互作用V为负值,吸收光谱红移,形成J-聚集;β大于32°时V为正值,形成H-聚集;当β=π/2时偶极相互作用为最大正值,为1 008 cm-1。
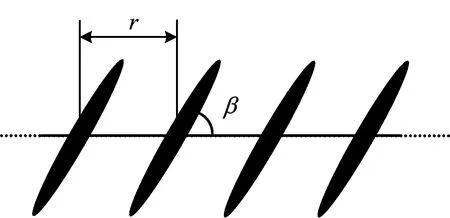
图4 叶黄素分子聚集体结构示意图
3.2.1 相互作用对聚集体吸收谱的影响
由计算可知当分子间相互作用越大,聚集体整体协作效应越明显,吸收光谱蓝移程度越大。图5是不同相互作用下的叶黄素H-聚集体的吸收光谱,模拟时取分子个数N=10,无序度D=0。图5中,V由小到大模拟所得的吸收谱的半高宽为:17,16和14 nm,远小于实验值的37 nm。

图5 不同相互作用情况下叶黄素聚集体的吸收谱模拟
图5还显示,当分子间相互作用V为1 000 cm-1时,10个叶黄素分子的聚集体吸收光谱蓝移至400 nm。当V增加到2 000 cm-1时,吸收谱蓝移至369 nm,与实验值大致接近。在聚集体模型中,分子间的最大正值偶极相互作用为1 008 cm-1,这不足以使吸收光谱产生77 nm的蓝移。因此分析认为当叶黄素分子形成紧密的H-聚集体后,分子间除了偶极相互作用外,还存在范德华力、疏水相互作用和氢键等,它们也会增加分子间的相互作用。假设所有的相互作用相加为2 000 cm-1,则模拟得到的吸收光谱的吸收峰在369 nm。可见在1∶1乙醇水溶液中,叶黄素聚集体分子间的相互作用大于1 000 cm-1。
3.2.2 分子个数对吸收光谱的影响
图6模拟了不同分子个数时的叶黄素H-聚集体吸收光谱,模拟时分子间相互作用取2 000 cm-1,无序度D=0。图6中当分子个数由5个增加到50个时,吸收光谱由376 nm移至365 nm,发生了11 nm的蓝移。同时还发现,随着分子个数的增加,吸收峰位也增加,但半高宽减小。图6中波长由大到小吸收峰的半高宽度依次是:20,10和6 nm,都小于图3实验吸收谱的37 nm。对于聚集体结构,单分子被光激发后,形成的激子不会局限在单分子上,而会扩展到更多的分子,形成聚集体整体的光响应。分子个数越多,分子激发态扩展范围越广,由于整体的协调效应及激发转移作用,使激子集中在某段较小的激发态能量区域,使分子聚集体的吸收谱线宽度往往比单分子吸收谱线宽度要小。
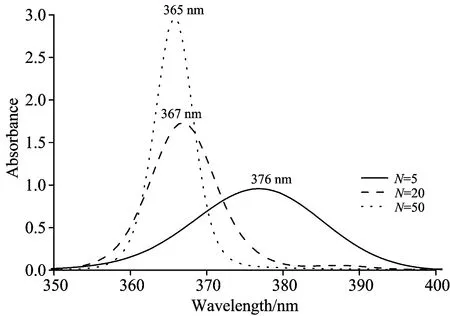
图6 不同分子个数情况下叶黄素聚集体的吸收谱模拟
3.2.3 无序度对吸收光谱的影响
由上述分析可知,相互作用和分子个数并不是理论模拟所得吸收光谱较实验值半高宽小的真正原因。分子聚集体通常由多个分子组成,每个分子的激发能受到环境影响会产生>随机的涨落,称为静态无序,用无序度D表示激发能偏离位点能的值。静态无序会减小聚集体的整体协同性,使分子吸收光谱产生非均匀展宽。图7模拟了10个分子的聚集体,分子间相互作用V=2 000 cm-1的情况下,不同无序度的吸收光谱。
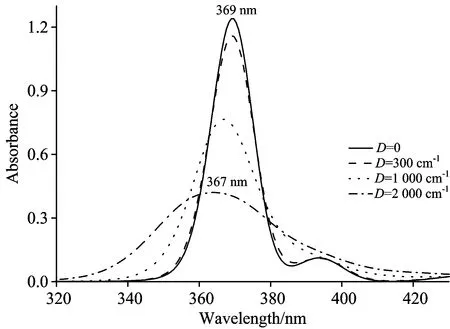
图7 不同无序度情况下叶黄素聚集体的吸收谱模拟
图7中无序度从小到大,吸收谱半高宽依次是:14,15,23和43 nm,吸收峰位从369移至367。可见随着无序度的增加,吸收谱线迅速展宽,峰强度下降,并发生微弱蓝移。由于D的增加,式(4)对角化得到的本征值分布变宽,能级分布可以上升到更高能量位置,粒子也能向更高能级跃迁,吸收波峰发生蓝移,这使得能级分布间隙变大,分子吸收不如原本那么集中,从而使吸收谱线展宽,随着吸收谱线宽度的增加,峰值强度下降。
4 结 论
实验中检测了叶黄素单体和H-聚集体的吸收光谱,并通过对叶黄素单体吸收光谱的高斯分解,获得了单分子振动能级的结构参数。理论上应用时间相关函数描述的吸收谱线和实验获得的结构参数,模拟了叶黄素单体的吸收谱线。采用Frenkel激子模拟了叶黄素H-聚集体的吸收光谱,分析了引起聚集体光谱变化的原因。结果显示,叶黄素H-聚集内由于分子间较强的相互作用(2 000 cm-1左右)使得吸收光谱发生蓝移,又由于协作效应峰形变窄。进一步考虑到分子个数和无序度的影响,吸收光谱的半高宽发生改变,模拟实验结果显示本实验中分子个数为10个~15个之间,无序度在1 000~2 000 cm-1之间 。研究结果为进一步探索聚集体的光谱行为和理解相关聚集系统功能提供了理论依据。
[1] Holt N E, Fleming G R, Niyogi K K. Biochemistry, 2004, 43: 8281.
[2] Chew B P, Park J S. J. Nutr., 2004, 134(1): 257S.
[3] Krinsky N I. J. Nutr., 2002, 132(3): 540S.
[4] Nikolay E P, Adam M, Lowell D K. J. Phys. Chem. B, 2013, 11: 10173.
[5] Hyesun P M, Jungsoo B, Samuel F L, et al. Biochimica et Biophysica Acta, 2007, 1768: 167.
[6] Polívka T, Sundström V. Chem. Rev., 2004, 104: 2021.
[7] Schulten K, Karplus M. Chem. Phys. Lett., 1972, 14: 305.
[8] Chen W, Christopher J B, Cheng H, et al. J. Phys. Chem. B, 2012, 116: 10617.
[9] Barvík I, Peineker P, Warns Ch, et al. Journal of Luminescence, 1995, 65: 169.
[10] LI Kai, HAN Jiao, ZHANG Hou-yu,et al(李 凯, 韩 蛟, 张厚玉,等). Chemical Journal of Chinese University(高等学校化学学报), 2011, 32(12): 2872.
[11] Shrieber Y, Toyozawa J. Phys. Soc. Japan, 1982, 51: 1528.
[12] CHEN Bao-jiu, WANG Hai-yu, HUANG Shi-hua(陈宝玖, 王海宇, 黄世华). Chinese Journal of Luminescene(发光学报), 2001, 22(3): 253.
[13] Ion A, Partali V, Sliwka H, et al. Electrochemistry Communications, 2002, 4(9): 674.
(Received Aug. 10, 2015; accepted Dec. 18, 2015)
*Corresponding author
Study on Absorption Spectra of Lutein Aggregate with Experimental Analysis and Theoretical Calculation
LU Li-ping,LI Ming,LIU Gui-ling,WEI Liang-shu,WU Fang*
College of Science, Nanjing Agricultural University, Nanjing 210095, China
Compared with the monomer, aggregate exhibits unique features such as electron and energy transfer which can be understood with the study of absorption spectra. In the experimental the absorption spectra of lutein monomer in ethanol solution and aggregate in 1∶1 aqueous ethanol solution are detected by utilizing UV-lVis spectrophotometer. The vibration structure of excited state of lutein monomer is obtained with Gauss decomposition of the absorption spectra. Theoretically, the molecular parameters of excitation energy, vibration frequency of characteristic mode, Huang-Phys factor are calculated by means of simulation of the monomer absorption spectra described by temporal correlation function and Frenkel exciton model. The spectral calculation of the lutein aggregate is conducted by using these parameters and then the factors of the spectral changes affected by the aggregate structure are analyzed. Some conclusions are drawn from the analysis: (1) The absorption peak position of the aggregate is determined mainly by intermolecular interaction. The calculation shows that the interaction is about 2 000 cm-1according 77 nm blue shift of absorption spectra from experiment result. (2) With the increase of molecular number of the aggregate the half-width of the absorption spectra decreases and the peak position blue shift slightly due to enhanced cooperation effect. (3) Disorder degree from environment has great influence on half-width for the greater disorder degree the larger half-width. The results of this paper will provide a theoretical reference for the further study of lutein aggregate function in biological and materials systems.
H-aggregate; Frenkel exciton; Absorption spectra; Cooperation effect
2015-08-10,
2015-12-18
中央高校基本科研业务费专项资金项目(KYZ201539,KYZ201425)资助
卢礼萍,女,1977年生,南京农业大学理学院讲师 e-mail: lplu0227@njau.edu.cn *通讯联系人 e-mail: wufang318@njau.edu.cn
O641
A
10.3964/j.issn.1000-0593(2016)10-3287-05
猜你喜欢
食品工业科技(2022年23期)2022-12-06
汽车实用技术(2022年14期)2022-07-30
河南工业大学学报(自然科学版)(2021年6期)2022-01-26
心理学报(2022年1期)2022-01-21
化工科技(2021年5期)2021-11-24
化工学报(2020年10期)2020-10-27
当代水产(2019年1期)2019-05-16
老年教育(老年大学)(2018年12期)2018-01-28
能源(2017年11期)2017-12-13
广东饲料(2016年4期)2016-12-01

- 光谱学与光谱分析的其它文章
- Gd靶激光等离子体光源离带辐射及其等离子体演化的研究
- Probing the Binding of Torasemide to Pepsin and Trypsin by Spectroscopic and Molecular Docking Methods
- Mn(Ⅱ)-5-Br-PADAP共沉淀-火焰原子吸收光谱法测定虾、贝样中的镉
- Near Infrared Spectroscopy Study on Nitrogen in Shortcut Nitrification and Denitrification Using Principal Component Analysis Combined with BP Neural Networks
- 内蒙古草原植被最大光能利用率取值优化研究
- 健康和糖尿病大鼠红细胞荧光光谱非线性程度差异