
Waardenburg综合征Ⅳ型患儿SOX10基因新突变研究
2016-06-30 09:06:02蔚开慧姜茜张震李颀肖萍苏琳邹继珍李龙潘尚领1广西医科大学基础医学院病理生理学教研室南宁300212首都儿科研究所遗传研究室北京1000203首都儿科研究所遗传研究室儿童发育营养组学北京市重点实验室北京100020首都儿科研究所附属儿童医院普通外科北京100020首都儿科研究所附属儿童医院病理科北京100020安徽医科大学合肥230001广西医科大学基础医学院病理生理学教研室南宁30021
中华耳科学杂志 2016年2期
蔚开慧姜茜张震李颀肖萍苏琳邹继珍李龙潘尚领1广西医科大学基础医学院病理生理学教研室(南宁30021)2首都儿科研究所遗传研究室(北京100020)3首都儿科研究所遗传研究室儿童发育营养组学北京市重点实验室(北京100020)首都儿科研究所附属儿童医院普通外科(北京100020)首都儿科研究所附属儿童医院病理科(北京100020)安徽医科大学(合肥230001)广西医科大学基础医学院病理生理学教研室(南宁30021)
Waardenburg综合征Ⅳ型患儿SOX10基因新突变研究
蔚开慧1,2姜茜3*
张震4李颀5肖萍5苏琳6邹继珍5李龙4潘尚领7
1广西医科大学基础医学院病理生理学教研室(南宁530021)
2首都儿科研究所遗传研究室(北京100020)
3首都儿科研究所遗传研究室儿童发育营养组学北京市重点实验室(北京100020)
4首都儿科研究所附属儿童医院普通外科(北京100020)
5首都儿科研究所附属儿童医院病理科(北京100020)
6安徽医科大学(合肥230001)
7广西医科大学基础医学院病理生理学教研室(南宁530021)
【摘要】目的通过分析研究一例Waardenburg综合征Ⅳ型(WS4)散发病例患儿的分子遗传学病因,丰富该致病基因突变谱,为WS4遗传咨询提供新的证据,并对该综合征相关的SOX10基因所有无义突变进行文献回顾和总结。方法收集一个WS4患儿的详细临床资料,签署知情同意书后获取血样,对包括SOX10、EDNRB、EDN3在内的172个先天性巨结肠及综合征相关基因进行二代测序,并用聚合酶链反应针对可疑致病突变进行扩增及Sanger测序验证,应用GeneTool软件及生物信息学网站的信息分析数据。结果发现患儿SOX10基因第4外显子存在一杂合无义突变(c.838G>T,p.E280X),父母均表现正常且未发现有该突变。结论发现一新的SOX10基因致病突变,丰富了致WS4的SOX10基因突变谱,并为父母提供再生育患儿的风险评估及必要的产前诊断咨询。
【关键字】Waardenburg综合征Ⅳ型;SOX10无义突变;目的基因测序;遗传咨询
蔚开慧和姜茜并列第一作者
No conflict of interest among the authors.
Waardenburg综合征(Waardenburg syndrome,WS)又称听力-色素综合征(auditory-pigmentary syndrome),是1951年由荷兰眼科遗传学医师PJ Waardenburg首先提出的一种较常见的综合征型遗传性耳聋,发病率约为1/42000,约占各类先天性耳聋的2%-5%[1,2]。WS被认为与胚胎期神经嵴细胞(neural crest cells,NCC)源性黑素细胞增殖、分化及迁移功能缺陷而导致的发育异常有关,以感音神经性聋、眼间距异常和虹膜、毛发、皮肤的色素沉淀异常为主要特征[3]。根据症状表现的不同可分为4个亚型:WS1主要表现为内眦外移易位(W>1.95)、鼻根宽阔、皮肤色素减退、听力障碍和虹膜异色症等;WS2 与WS1相比无内眦外移易位(W<1.95)、鼻根宽阔的表现;WS3与WS1的表型相似,其显著特点为同时伴颜面或上肢肌肉骨骼的发育异常;WS4与WS2表型相似但伴有先天性巨结肠[2,4,5]。
截至目前,20%-40%的WS4患者发病原因不明,另有约20%-30%的患者由EDN3或EDNRB基因突变引起,绝大多数WS4患者(约45%-55%)均是由SOX10突变所引起,且无义突变居多[6]。SOX10是NCC迁移分化过程中的一种关键的转录因子,主要通过与靶基因(如MITF、TYR、TYRPl、DCT)的增强子或启动子结合来发挥其转录调节功能。1998年Pingault V等首次在WS4患者中发现了SOX10基因的杂合突变,确定SOX10为WS4的第三个致病基因,并进一步研究证实该基因所编码蛋白对小鼠及人神经嵴发育的重要调节作用[7,8]。后又发现该基因突变可导致另一种伴有严重神经系统疾病的综合征PCWH (peripheral demyelinating neuropathy,central dysmyelinating leukodystrophy,WS,and Hirschsprung disease)[9]。此后,在WS4患者中检测到SOXl0基因突变的报道越来越多,Lu Jiang[10]等于2011年首次报道了2例来自中国的WS4伴SOX10基因突变(c.254G>A 和c.698-2A>T)的患儿,也证实了突变基因SOX10与WS4的相关性。但目前发现的与WS4/PCWH相关的SOX10突变中只有3个来自中国,且突变所致的表型高度变异,还需要更多的相关报道来完善。
本研究收集并分析一WS4散发病例患儿,发现先证者携带的SOX10基因杂合无义突变(c.838G>T,p.E280X,E4)国内外尚未见报道,属新突变,且患儿父母均正常无突变,该突变为新发突变。我们进一步对文献已报道的所有WS4相关SOX10基因无义突变病例的临床表型进行了总结,希望探索该综合征表型与基因型之间可能存在的关系。
1 材料与方法
1.1临床资料
先证者及其父母来自中国大连,问卷式采集先证者及其父母的健康资料,并进行耳科、眼科、皮肤、毛发、消化系统和四肢关节等全面体格检查。取得家庭成员的知情同意并签署知情同意书后获取先证者及其父母的外周静脉血各5ml,用于基因组DNA的提取。
1.2172个先天性巨结肠及综合征相关基因二代测序
(1)DNA提取及全基因组文库制备:应用Qiagen公司试剂盒(the QIAamp DNA Blood Midi Kit,Qiagen,Hilden,Germany)提取患儿静脉血基因组DNA,将质量检测合格的基因组DNA随机打断(Biorupter),纯化长度在150~250bp之间的片段;然后利用T4 DNA Polymerase、T4 phosphorylated polynucleotide kinase和Eseheriehia coli DNA聚合酶Klenow片段对纯化后的DNA片段进行末端修复,再按照Illumina公司二代测序仪的操作说明在片段两端加上A碱基,最后在两端加上接头(adaptor)并对其进行磁珠纯化。
(2)杂交:已加接头纯化后的模板首先进行捕获前PCR扩增,然后将PCR产物与自主设计的GenCap Custom Enrichment Kit(北京迈基诺基因科技有限责任公司)在适宜条件下杂交22h,此时目标片段因与特异性探针结合而被捕获,未杂交的片段则被洗掉,洗脱后保留在探针上的目标片段再进行一次捕获PCR以显著增加待测片段的数量。
(3)生物信息学分析:本实验中使用的芯片捕获区包含172个先天性巨结肠及综合征相关基因的外显子及其侧翼序列约100 bp。高通量测序数据使用Illumina Pipeline(version 1.8.2)产生原始数据,去除低质量的数据后利用BWA(Burrows wheeler aligner)将“干净的”读序与人类基因组参考序列比对(UCSC,hg19),再分别使用SOAPsnp软件和GATK软件进行SNP和InDel的收集。本实验中样品基因的平均测序深度约为96×,捕获区覆盖度达90%以上,为了找出致病性的点突变,参考dbSNP数据库、Hapmap、千人基因组数据库及ESP(NHLBI Exome Sequencing Project)和内部正常对照人群数据库,将频率小于0.05的变异视为可疑。
1.3先证者及其父母突变验证
应用在线PRIMER3对可疑突变(c.838G>T)设计引物:正向引物:5'-ccttgcgctctctctctctg-3',反向引物:5'-GGCAGGTACTGGTCCAACTC-3',扩增片段长度为244bp。采用盐析法提取患儿及其父母的静脉血基因组DNA并进行PCR扩增,反应体系(50μl)包含10ng/μl DNA模板15μl,20μM正、反向引物各2μl,10×PCR buffer 5μl,2.5mM dNTPmix 5μl,Takara Taq酶0.5μl,去离子水20.5μl。反应条件为94℃预变性1min,35个循环(94℃变性30s,55℃退火30s,68℃延伸30s),72℃补充延伸1min,将扩增产物保存在4℃。PCR扩增产物经纯化及双向测序后(北京诺赛基因)进行突变分析。
2 结果
2.1临床资料分析患儿,女,出生后2天听力筛查及42天复筛双侧耳声发射均未通过,双耳听力诱发电位检查异常。双侧虹膜呈浅蓝色,生后2个月即因确诊先天性巨结肠长段型行横结肠造瘘术。临床拟诊为Waardenburg综合征IV型。患儿足月顺产,出生体重2950g,身长49cm,出生时反应尚可,Apgar评分9分-9分-9分。患儿于出生6个月时突然死亡,原因不明,患儿父母均表现正常,无家族史。
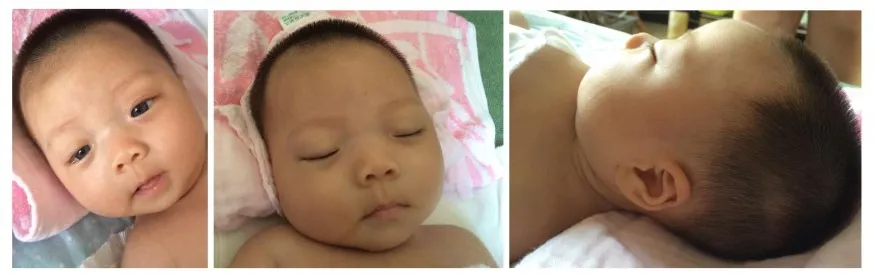
图1 患儿面部正侧位照片。双眼虹膜着色异常,呈蓝色,余未见明显异常。头围正常。皮肤、毛发颜色正常。Fig.1 Anterior and lateral photographs of the patient's face.Her irises were in light blue and no other abnormalities were observed.Her head circumference was in the normal range,as with the color of skin and hair.
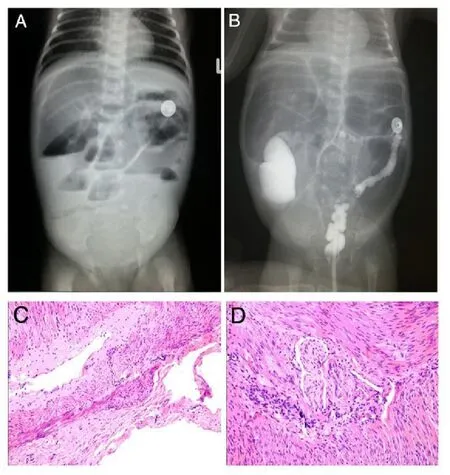
图2 患儿影像学表现及术中病理切片鉴别诊断(HE染色)。A:腹部X线平片提示为肠梗阻表现,盆腔无气。B:腹部下消化道造影显示直肠、乙状结肠、降结肠、横结肠痉挛狭窄,升结肠、盲肠扩张。小肠胀气明显。C:患者术中横结肠远端冰冻切片普通HE染色×200,D:患者术中乙状结肠冰冻切片普通HE染色×200,两张切片均未找见神经节细胞,证实为长段型先天性巨结肠。Fig.2 Imaging findings and pathological diagnosis of the patient in operation(HE staining).A:Abdominal X-ray plain film of the patient showed intestinal obstruction and pelvic airless.B:Lower intestinal radiography of the patient showed spasm narrow in the rectum,sigmoid colon,descending colon,transverse colon and expansion in the ascending colon,the cecum.Small intestine bloated severely.C:The distal transverse colon frozen section of the patient in operation showed a long segment aganglionisis(×200 magnification,HE),D:The sigmoid colon frozen section of the patient in operation showed a long segment aganglionisis(×200 magnification,HE).
2.2突变分析
对患儿进行172个先天性巨结肠及综合征相关基因二代测序后发现,3个WS4已知致病基因中只有SOX10在第4外显子存在杂合无义突变(c.838G>T,p.E280X,表1),且该突变国内外均尚未见报道,为一新突变。对患者父母进行突变验证后证实此突变为新发突变(见图3 A)。
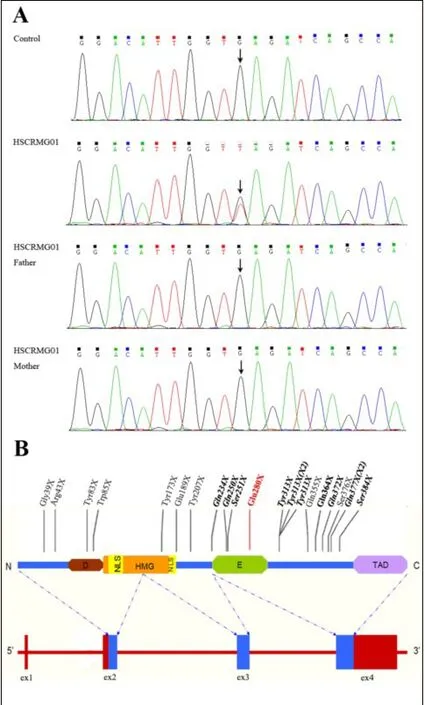
图3 患儿及其父母SOX10基因测序结果及模式图。A:对照(control)、患者(HSCRMG01)、患者父亲(HSCRMG01 Father)及患者母亲(HSCRMG01 Mother)的Sanger测序结果,箭头所示为c.838G>T突变。B:SOX10基因(下)及所编码蛋白(上)的模式图,图中D代表二聚体化功能域;NLS代表核定位信号;HMG代表高度保守的高活性组分结构域;E代表SOX8/9/ 10的一个保守区域;TAD代表C端转录激活域。与PCWH相关的无义突变用黑色加粗字体表示;与WS4相关的无义突变用黑色字体表示;本研究新发现的无义突变标记为红色字体。×2表示同一突变报道2次。Fig.3 Sequencing results and schematic representation of the SOX10 mutation.A:Sanger sequencing chromatogram of the SOX10 mutation from the control,the patient(HSCRMG01)and her parents,the arrow indicates the site of the mutation (c.838G>T,p.E280X).B:Nonsense mutations identified in the schematic representation of SOX10 gene(below)and protein (above).“D”represents a dimerization domain;“NLS”represents nuclear localization signal;“HMG”represents a highly conserved DNA-binding high mobility group domain;“E”represents a conserved domain in SOX8/9/10;“TAD”represents a carboxyterminal transactivation domain.Mutations associated with PCWH are in bold,mutations associated with WS4 are in black,and the novel mutation founded in our study is in red.The“×2”indicates one mutation has been reported twice.
2.3SOX10基因无义突变文献回顾与总结
结合近年文献报道,总结发现导致WS4或PCWH的SOX10无义突变共有20种(WS4和PCWH各有10种),约占导致WS4或PCWH的所有SOX10突变的44%。可见,SOX10无义突变在WS4和PCWH的发生中均扮演了重要角色,具体无义突变信息总结见图3 B和表2。从表2可见所有突变均为杂合突变,男性多发,且先证者合并其他突变(如EDNRB、EDN3)的可能性很小。同时还发现绝大多数已报道的与WS4相关的SOX10无义突变均为新发突变,只有4例有明确的家族遗传倾向(p.W85X、p.Y313X、p.Q377X和p.S376X,见表2),p.W85X、p.Y313X和p.Q377X先证者父母中均有一位表型正常的杂合突变携带者,p.S376X患儿的母亲有耳聋的症状,基因型未知,p.W85X和p.Q377X先证者家系中均有一位携带杂合突变的姊妹患病。从图3 B中则可看出引起短段型巨结肠或单纯型WS4的无义突变多发生在2、3外显子上,引起长段型、全结肠型巨结肠或PCWH的无义突变多发生在第4外显子上,而TAD域内极少发生突变。
2.4再生育聋儿风险评估
鉴于患儿父母均表现正常,无相应突变,对该家庭进行再生育聋儿风险评估可分为两种情况:如果其父母中有一方为生殖细胞镶嵌体,再生育后代发生该基因杂合突变的风险为50%,可以通过产前SOX10基因同位点测序进行明确的产前诊断;如果父母的体细胞和生殖细胞均无突变,则再生育后代发生相同突变且患病的可能性极小(约相当于该病的一般人群发病率1/42000),但是也建议他们进行产前SOX10、EDNRB、EDN3基因全序列检查以明确胎儿的基因型,若基因诊断结果显示有突变则再生育后代为一聋儿的风险大于90%(由表2总结:有突变而表型正常者约占突变携带者总人数的10%)。
3 讨论
SOXl0基因(性别决定区盒基因,SRY - box 10)位于22q13,是神经系统发育中的一种重要的转录因子,含有4个外显子,其中只有2、3、4号外显子编码蛋白(见图3 B)。SOX10最先在NCC迁移早期的胚胎背神经管中表达[11],然后随着NCC的逐渐分化而在其衍生物如黑素细胞、胃肠道系统及周围神经系统中表达。SOX1O蛋白属于20个SOX基因超级家族成员中的一员,含有466个氨基酸,相对分子质量约为51kDa,发挥主要功能的是高活性组分结构域(high mobility group,HMG)和C端转录激活域(C-terminal transactivation domain,TAD)[12]。HMG域由80个氨基酸(第102-181位)组成,形成3个“L”型弯曲排列的α螺旋区,可单独或与其他转录因子协同作用来识别靶基因特异DNA序列并与其小沟结合,导致DNA的构象发生改变,TAD域可激活该基因的转录[13]。研究发现SOX10可通过与MITF启动子中特定序列结合而使MITF的转录活性提高100倍,且SOX10与PAX3的协同作用可增强这种激活效应[14,15]。HMG域的两侧各有一个核定位信号(nuclear localization signal,NLS),可使SOX10蛋白在细胞质与细胞核之间穿梭运动,进而可能影响黑素细胞或神经嵴的发育[16]。
SOX10基因突变曾先后在WS2和WS4/PCWH患者中发现过,已被确定为这两种亚型的致病基因。截至2014年,文献已报道的SOX10基因突变共有82种(http://grenada.lumc.nl/LOVD2/WS/home.php),其中绝大多数为碱基置换突变(46种)和缺失突变(23种),在突变蛋白中无义突变和移码突变均有25种,错义突变有19种。文献已报道的与疾病相关的SOX10突变共有66种,其中与WS4或PCWH相关的有45种(WS4有22种,PCWH有23种),与WS2相关的只有7种(http://research.nhgri.nih.gov/pigment_cell/ templates/files/SOX10_polymorphism.shtml)。SOX10所引起的WS4/PCWH的遗传方式主要为单基因致病的常染色体显性遗传伴不全外显,而EDNR3、EDNRB所引起的WS4则主要表现为常染色体隐性遗传。Inoue等[17]发现当无义突变发生在第4外显子以前时会激活无义介导的mRNA降解通路(nonsense-mediated mRNA decay,NMD),突变mRNA会被识别、降解,导致野生型等位基因编码的蛋白质的总量不足(即单倍体剂量不足效应,haploinsufficiency),从而造成杂合突变基因所编码的蛋白质无法发挥正常的生理功能,引起WS4。当无义突变发生在第4外显子时,突变mRNA不仅不会被降解,而且还会通过显性负效应竞争性结合DNA抑制野生SOX10蛋白功能而导致严重的WS4表型或PCWH。Honghan Wang[18]通过实验发现突变型蛋白(c.1063C>T,p.Q355X,E4)的表达量及与DNA结合的能力与野生型相似,但随着突变蛋白表达量的增加,其下游基因MITF的表达量逐渐下降。Sham et al.[19]发现发生在SOX10上游的无义突变常引起较轻的短段型巨结肠,而发生在SOX10最后一个外显子上的无义突变常引起较严重的神经节细胞缺乏症,这种趋势也可以从表2所总结的临床表现中观察出来。Pingault[20]等报道的无义突变(p.R43X)发生在该基因的上游,先证者表现为较轻的短段型巨结肠。突变p.Gln234X则导致严重的PCWH表型,为目前发现未被降解的最短的截短蛋白。更多的基因型-表型关联还需要进一步扩大患者样本量继续研究。
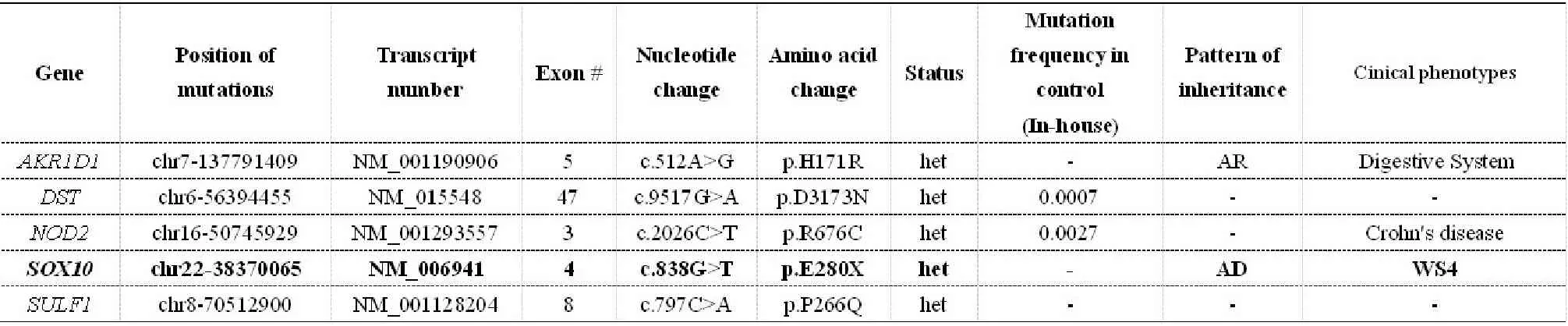
表1 患儿(HSCRMG1)172个目的基因测序可疑突变位点检测结果Table 1 Sequencing results of the suspected mutations from 172 related genes in patient HSCRMG1
本次研究中,本研究在一散发WS4先证者中发现了一个新的杂合无义突变(c.838G>T,p.E280X,E4),该突变位于高度保守的SOX Group E结构域内,即HMG域与TAD域之间。突变使第280位的谷氨酸变为一个提前终止的密码子,所产生的截短蛋白由于TAD域的缺失而失去了对下游靶基因启动子或增强子的转录激活功能,从而可能影响神经嵴细胞和黑素细胞的胚胎期发育,产生WS4的一系列症状。患儿于生后6个月时突然死亡,死因不明,同时我们可以从表2中发现位于SOX Group E结构域内的4个无义突变的患儿均在婴儿期死亡,可以推测SOX Group E结构域可能与某种严重危及生命的机体功能相关,有待进一步研究。
本研究不仅对患者及其家庭进行了基因诊断和风险评估,还对该综合征相关的SOX10基因所有无义突变进行了文献回顾和总结。新突变位点丰富了相关综合征的疾病突变数据库,同时还为深入了解SOX10基因型-表型关联提供了新的证据。
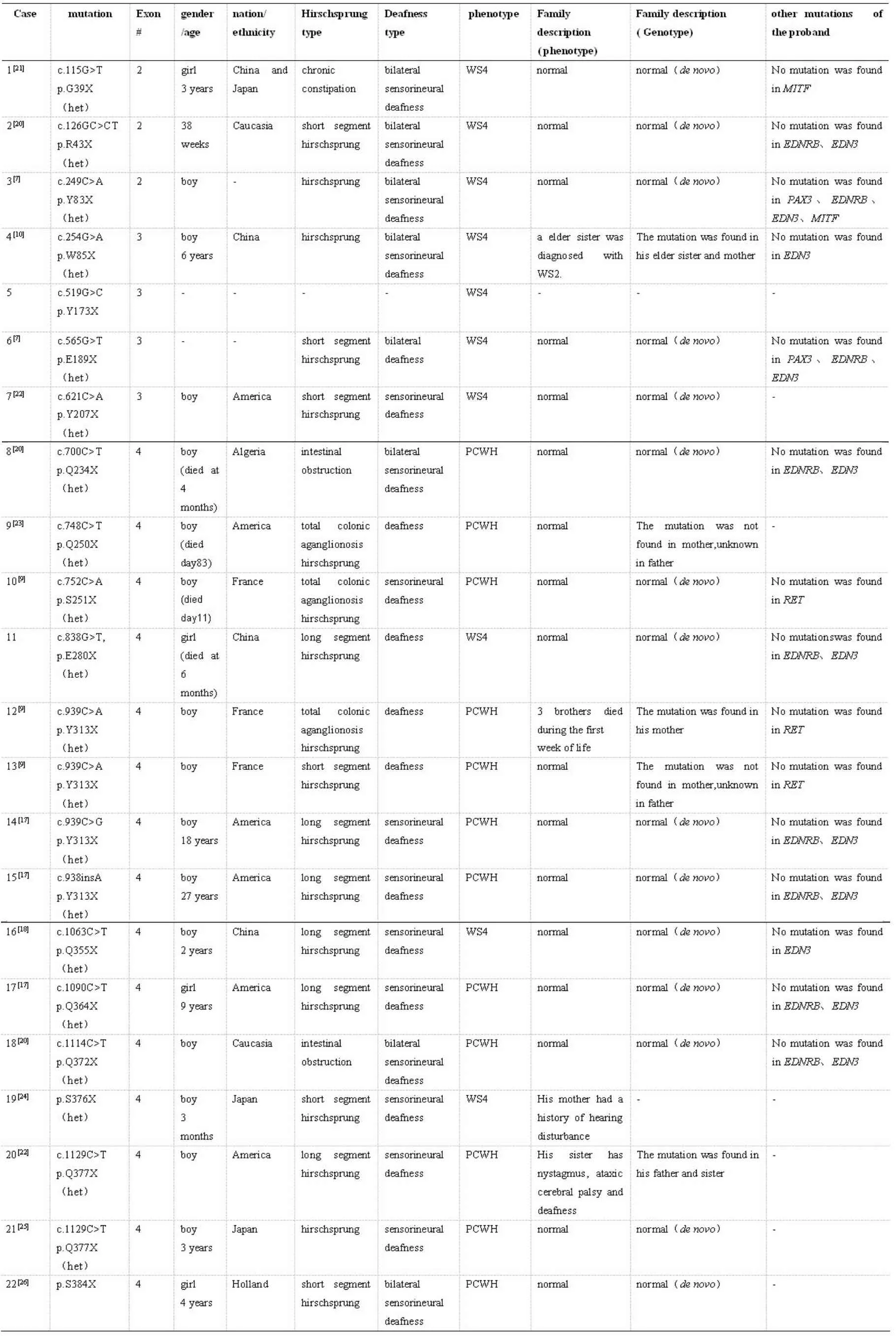
表2 已报道的WS4或PCWH表型患者的SOX10基因无义突变总结Table 2 Reported cases of SOX10 nonsense mutations associated with WS4 or PCWH
参考文献
1Waardenburg PJ.A new syndrome combining developmental anomalies of the eyelids,eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness.Am J Hum Genet,1951,3(3):195-253.
2Read AP,Newton VE.Waardenburg syndrome.J Med Genet,1997,34(8):656-665.
3Baxter LL,Hou L,Loftus SK,et al.Spotlight on spotted mice:a review of white spotting mouse mutants and associated human pigmentation disorders.Pigment Cell Res,2004,17(3):215-224.
4Pingault V,Ente D,Dastot-Le Moal F,et al.Review and update of mutations causing Waardenburg syndrome.Hum Mutat,2010,31 (4):391-406.
5高紫璇,李金红,卢宇,等.Waardenburg综合征Ⅱ型一家系基因突变研究.中华耳科学杂志,2014,12(2):271-271.GAO Zixuan,LI Jinhong,LU Yu,et al.A Genetic Mutation Study in a Pedigree with Type II Waardenburg Syndrome.Chinese Journal of Otology,2014,12(2):271-271.
6Bondurand N,Dastot-Le Moal F,Stanchina L,et al.Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4.Am J Hum Genet,2007,81(6):1169-1185.
7Pingault V,Bondurand N,Kuhlbrodt K,et al.SOX10 mutations in patients with Waardenburg-Hirschsprung disease.Nat Genet,1998,18(2):171-173.
8Herbarth B,Pingault V,Bondurand N,et al.Mutation of the Sry-related Sox10 gene in Dominant megacolon,a mouse model for human Hirschsprung disease.Proc Natl Acad Sci U S A,1998,95(9):5161-5165.
9Touraine RL,Attie-Bitach T,Manceau E,et al.Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain.Am J Hum Genet,2000,66(5):1496-1503.
10 Jiang L,Chen H,Jiang W,et al.Novel mutations in the SOX10 gene in the first two Chinese cases of type IV Waardenburg syndrome.Biochem Biophys Res Commun,2011,408(4):620-624.
11Southard-Smith EM,Kos L,Pavan WJ.Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model.Nat Genet,1998,18(1):60-64.
12 Harris ML,Baxter LL,Loftus SK,et al.Sox proteins in melanocyte development and melanoma.Pigment Cell Melanoma Res,2010,23 (4):496-513.
13 Kuhlbrodt K,Schmidt C,Sock E,et al.Functional analysis of Sox10 mutations found in human Waardenburg-Hirschsprung patients.J Biol Chem,1998,273(36):23033-23038.
14Bondurand N,Pingault V,Goerich DE,et al.Interaction among SOX10,PAX3 and MITF,three genes altered in Waardenburg syndrome.Hum Mol Genet,2000,9(13):1907-1917.
15 Potterf SB,Furumura M,Dunn KJ,et al.Transcription factor hierarchy in Waardenburg syndrome:regulation of MITF expression by SOX10 and PAX3.Hum Genet,2000,107(1):1-6.
16 Sinner D,Kordich JJ,Spence JR,et al.Sox17 and Sox4 differentially regulate beta-catenin/T-cell factor activity and proliferation of colon carcinoma cells.Mol Cell Biol,2007,27(22):7802-7815.
17 Inoue K,Khajavi M,Ohyama T,et al.Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations.Nat Genet,2004,36(4):361-369.
18 Wang HH,Chen HS,Li HB,et al.Identification and functional analysis of a novel mutation in the SOX10 gene associated with Waardenburg syndrome type IV.Gene,2014,538(1):36-41.
19 Sham MH,Lui VC,Chen BL,et al.Novel mutations of SOX10 suggest a dominant negative role in Waardenburg-Shah syndrome.J Med Genet,2001,38(9):E30.
20Pingault V,Girard M,Bondurand N,et al.SOX10 mutations in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism.Hum Genet,2002,111(2):198-206.
21 Arimoto Y,Namba K,Nakano A,et al.Chronic constipation recognized as a sign of a SOX10 mutation in a patient with Waardenburg syndrome.Gene,2014,540(2):258-262.
22 Southard-Smith EM,Angrist M,Ellison JS,et al.The Sox10(Dom)mouse:modeling the genetic variation of Waardenburg-Shah(WS4)syndrome.Genome Res,1999,9(3):215-225.
23 Inoue K,Shilo K,Boerkoel CF,et al.Congenital hypomyelinating neuropathy,centraldysmyelination,andWaardenburg-Hirschsprung disease:phenotypes linked by SOX10 mutation.Ann Neurol,2002,52(6):836-842.
24 Toki F,Suzuki N,Inoue K,et al.Intestinal aganglionosis associated with the Waardenburg syndrome:report of two cases and review of the literature.Pediatr Surg Int,2003,19(11):725-728.
25 Oshimo T,Fukai K,Abe Y,et al.Pediatric case report:clinical profile of a patient with PCWH with p.Q377X nonsense mutation in the SOX10 gene.J Dermatol,2012,39(12):1022-1025.
26 Verheij JB,Sival DA,van der Hoeven JH,et al.Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations:a case report and review of the literature.Eur J Paediatr Neurol,2006,10(1):11-17.
Novel SOX10 mutation in a girl with type IV Waardenburg syndrome
YU Kaihui1,2JIANG Qian3*,ZHANG Zhen4,LI Qi3,XIAO Ping5,SU Lin6,ZOU Jizhen5,LI Long4,PAN Shangling7
1 Department of Pathophysiology,School of Preclinical Sciences,Guangxi Medical University,Nanning,530021
2 China;Department of Medical Genetics,Capital Institute of Pediatrics,Beijing,100020
3 Department of Medical Genetics,Beijing Municipal Key Laboratory of Child Development and Nutriomics,Capital Institute of Pediatrics,Beijing,100020
4 Department of General Surgery,The Children's Hospital Affiliated to Capital Institute of Pediatrics,Beijing,100020
5 Department of Pathology,The Children's Hospital Affiliated to Capital Institute of Pediatrics,Beijing,100020
6 Anhui Medical University,Hefei,230001,China
7 Department of Pathophysiology,School of Preclinical Sciences,Guangxi Medical University,Nanning,530021
Corresponding author:PAN ShanglingEmail:slpan@gxmu.edu.cn
【Abstract】Objective To improve the disease causative gene mutation spectrum and provide new information for genetic counseling in Waardenburg syndrome type IV(WS4),we performed molecular genetic study in an isolated patient affected by WS4.Literature was reviewed for reported nonsense mutations in SOX10.Methods Detailed histories were collected through questionnaires and physical examination.Blood samples of the patient and her parents were collected after obtaining informed consents.Suspected mutations were amplified and verified by Sanger sequencing after the next generation sequencing of 172 related genes.The raw data were analyzed using molecular biological websites and the GeneTool software.Results A new de novo heterozygous mutation(c.838G>T,p.E280X)in the fourth exon of SOX10 was found inthe patient.Both parents were demonstrated to be wild-type and symptom free.Conclusions The novel mutation found in our study not only enriches the mutation spectrum but also is helpful for recurrent risk evaluation and genetic counseling for this family.
【Key words】Waardenburg syndrome typeⅣ;SOX10 nonsense mutation;Target gene sequencing;Genetic counseling
【中图分类号】R764
【文献标识码】A
【文章编号】1672-2922(2016)02-240-7
DOI:10.3969/j.issn.1672-2922.2016.02.023
*基金项目:国家自然科学基金青年科学基金项目(81300266);北京市自然科学基金面上项目(7142029);北京市优秀人才培养个人项目(2013D003034000007);北京市科技新星(Z151100000315091)
作者简介:蔚开慧,硕士研究生,研究方向:先天性巨结肠的分子和遗传学发病机制
通讯作者:潘尚领,Email:slpan@gxmu.edu.cn
收稿日期:(2015-12-21审核人:袁永一)
*Fund projects:National Natural Science Foundation of China(No.81300266),the Beijing Natural Science Foundation(No.7142029),the Beijing Excellent Scientist Fund(No.2013D003034000007)and Beijing Novo Program(Z151100000315091)
猜你喜欢
青年文学家(2024年10期)2024-05-26 18:27:59
北京大学学报(医学版)(2022年2期)2022-11-21 03:29:12
临床输血与检验(2022年3期)2022-06-22 02:52:50
种子(2021年3期)2021-04-12 01:42:22
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
遗传(2019年5期)2019-05-21 09:58:28
传染病信息(2019年2期)2019-05-17 13:16:04
岷峨诗稿(2018年2期)2018-11-14 18:51:09
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
重庆医学(2015年12期)2015-03-05 05:52:54
