
肿瘤分子靶向治疗的研究进展
2016-06-20 05:29杜鲁巴孙浩然综述钦伦秀审校
复旦学报(医学版) 2016年1期
张 钰 杜鲁巴 孙浩然(综述) 钦伦秀,3△(审校)
(1复旦大学附属华山医院普外科 上海 200040; 2复旦大学肿瘤转移研究所 上海 200040;3复旦大学生物医学研究院 上海 200032)
肿瘤分子靶向治疗的研究进展
张钰1,2杜鲁巴1,2孙浩然1,2(综述)钦伦秀1,2,3△(审校)
(1复旦大学附属华山医院普外科上海200040;2复旦大学肿瘤转移研究所上海200040;3复旦大学生物医学研究院上海200032)
【摘要】近十几年来分子靶向治疗在血液系统及实体肿瘤中均取得较好的临床疗效。然而,不可避免发生的耐药现象已经成为靶向治疗的瓶颈。随着对分子靶向治疗药物耐药机制的深入研究,逐步发展逆转和预防耐药的策略。本文着重总结肿瘤分子靶向治疗的临床应用及关键临床试验、靶向治疗耐药机制以及临床中有效的药物联合治疗策略等方面的进展。
【关键词】肿瘤;靶向治疗;耐药;联合用药
恶性肿瘤严重威胁人类生命,其发病率及死亡率逐年上升。2012年全世界共新增1 400万癌症病例并有820万人死亡[1]。自20世纪40年代细胞毒性物质氮芥用于治疗淋巴瘤开始,近几十年大量的抑制DNA合成及功能的细胞毒性药物被合成并用于临床,成为目前临床中应用最广泛的抗肿瘤药物[1]。但是细胞毒性药物除了对睾丸癌及淋巴瘤等具有较好的治疗效果外,并不能明显改善大多数其他肿瘤患者的预后,并且细胞毒性药物特异性差,使细胞毒性药物的发展进入了一个瓶颈期。近年来随着分子生物学及遗传学等研究的进展,人们发现恶性肿瘤表现出复杂的特异性的生物缺陷,包括癌基因、抑癌基因突变及染色质修饰等。肿瘤分子靶向治疗就是在此基础上,利用肿瘤组织或细胞所具有的特异性结构分子为靶点,使用能与这些靶分子特异性结合的药物,特异性地杀伤肿瘤细胞的治疗[2]。随着靶向治疗药物取得较好的临床治疗效果,肿瘤分子靶向治疗成为近年来肿瘤治疗的研究热点。但是尽管肿瘤的分子靶向治疗进展迅速,其原发性或获得性耐药却限制了其临床应用。如何克服靶向治疗的缺陷、合理地联合用药从而改善患者预后成为了人们面临的巨大挑战。本文就近年来肿瘤分子靶向治疗药物的临床应用、耐药机制及联合用药策略作一综述。
分子靶向药物的临床应用
过去的十几年,多个针对不同癌基因信号通路的靶向治疗药物上市,引发了癌症治疗的革命。2001年第一个小分子BCR-ABL激酶抑制剂甲磺酸伊马替尼 (Gleevec)上市治疗Ph+慢性髓细胞淋巴瘤,开创了靶向激酶信号通路肿瘤治疗的先河。2003年吉非替尼 (Iressa)成功用于具有表皮生长因子受体 (EGFR) 基因突变的晚期非小细胞肺癌优势人群,开启了基于生物标志物的个体化治疗新时代。2004 年第一个抑制新生血管生成的VEGFR单克隆抗体贝伐单抗 (Bevacizumab)问世用于结肠癌治疗,开启了靶向肿瘤新生血管治疗的序幕。随后在短短的几年间,先后涌现出针对肿瘤细胞增殖信号转导通路的克唑替尼、厄罗替尼和威罗菲尼以及针对肿瘤新血管生成的血管内皮抑素、索拉非尼及苏尼替尼等多个分子靶向药物,并在不同的肿瘤中取得了较好的临床治疗效果。近年来研究发现肿瘤长期处于慢性应激状态,因而调控细胞染色质修饰的蛋白、分子伴侣及泛素-蛋白酶体等对肿瘤细胞的增殖和存活比正常细胞更为重要,由于这些分子与肿瘤细胞的DNA复制及分裂等直接相关,针对这些分子的靶向治疗研究逐渐兴起,随着蛋白酶体抑制剂硼替佐米在多发性骨髓瘤的治疗中取得较好的疗效,开启了分子靶向治疗的新方向。以下将对重要的肿瘤靶点及其靶向药物进行叙述(表1)。
靶向HER2治疗乳腺癌约25%~30%的乳腺癌患者存在HER2基因过表达,HER2 通路在乳腺癌发生、发展中起到非常重要的作用[3]。曲妥珠单抗是首个被批准上市的靶向HER2的治疗药物。两项Ⅲ期临床研究表明曲妥珠单抗联合紫杉醇或多西他赛治疗与单药化疗相比,能够给晚期转移性乳腺癌患者带来明显的生存获益[4-5]。基于这两项研究结果,曲妥珠单抗确立了其在晚期乳腺癌标准治疗的重要地位。曲妥珠单抗更令人瞩目之处在于其在术后预防复发转移的辅助治疗领域取得了“革命性”的结果。2005年多个国际多中心随机对照研究证实曲妥珠单抗用于早期乳腺癌术后辅助治疗1年,能使HER2+乳腺癌患者复发风险下降39%~52%[6-7]。尼洛替尼(Neratinib)是一种不可逆的泛HER受体酪氨酸激酶抑制剂,近期的一项II期临床实验表明在HER2+的进展期乳腺癌患者使用曲妥珠单抗后再用Neratinib治疗,其客观反应率 (objective response rate,ORR)为24%,中位无疾病进展生存期 (median progression-free survival,PFS)为22周,而未使用曲妥珠单抗组的ORR及中位PFS明显较高分别为56%及39.6周,Neratinib在治疗进展期 HER2 阳性乳腺癌方面的展现出了较好的治疗效果。
靶向KIT及PDGFRA治疗胃肠道间质瘤胃肠道间质瘤(gastrointestinal stromal tumors,GIST) 是胃肠道最常见的间叶组织源性肿瘤,约92%~95%的GIST患者存在干细胞因子受体 (KIT) 染色阳性,此外约5%~7% GIST患者存在血小板源性生长因子受体 (PDGFRA)基因变异。KIT和PDGFRA基因突变激活被认为与超过90%的恶性GIST 发生发展密切相关[8]。甲磺酸伊马替尼是第一个基于肿瘤细胞信号转导机制的认识而开发的小分子酪氨酸激酶受体抑制剂,能够选择性地靶向抑制KIT和PDGFRA等靶点。多个Ⅲ期临床研究表明甲磺酸伊马替尼能够使70%~85%的晚期GISTs患者的中位PFS延长到20~24个月,并使晚期GIST患者的中位总生存期延长至36个月[9-10]。尼洛替尼是一种新型的抑制KIT、PGDGR及BCR-ABL的酪氨酸激酶的抑制剂,2012年的一项Ⅲ期临床实验表明尼洛替尼可以明显提高伊马替尼及苏尼替尼耐药或不耐受的晚期GIST患者的总生存期 (overall survival,OS)[11]。但是2015年的一项Ⅲ期临床实验对比了伊马替尼及尼洛替尼作为一线药物治疗GIST的效果,发现相较于尼洛替尼,伊马替尼可以明显提高转移或无法手术切除患者的PFS[12]。
靶向BRAF治疗恶性黑色素瘤恶性黑色素瘤患者约60%存在BRAF基因突变,且90%的BRAF突变为持续活化的V600E基因突变[13]。一项Ⅲ期临床研究表明BRAF-V600E基因突变体抑制剂维罗非尼(Vemurafenib)对80%的BRAF-V600E基因突变的恶性黑色素瘤患者有效[14]。维罗非尼也成为第一个FDA批准的可延长BRAF-V600E基因突变阳性转移性黑色素瘤患者生存期的靶向药物。
靶向EGFR治疗非小细胞肺癌(NSCLC)NSCLC存在多个驱动基因,如EGFR基因活化突变、K-Ras基因突变和EML4 /ALK基因的表达增加等。研究表明约43% ~ 89%的NSCLC患者肺癌组织标本中检测到EGFR基因突变[15]。厄洛替尼及吉非替尼是一类是小分子EGFR-TKIs,通过竞争性结合EGFR酪氨酸激酶区域的ATP结合位点,抑制其酪氨酸激酶活性。多个Ⅲ期临床研究奠定了EGFR-TKIs在EGFR突变型NSCLC中的一线治疗地位,治疗后患者PFS可达9~13个月[16-17]。EGFRT790M基因突变的患者往往对吉非替尼及厄洛替尼治疗耐药,近期的一项临床试验检测了针对EGFRT790M基因突变的靶向药物AZD9291对127例EGFRT790M基因突变患者的治疗效果,结果表明61%的患者接受了AZD9291治疗之后出现明显的肿瘤缩小[18]。
靶向BCR-ABL治疗慢性粒细胞性白血病(CML)CML患者90% 以上骨髓细胞中存在特征性的费城染色体,其基因型为bcr-abl,是第9号染色体上的abl原癌基因与第22 号染色体上的bcr基因相互易位形成的融合基因,可引起蛋白激酶持续性激活。甲磺酸伊马替尼是靶向BCR-ABL的小分子TKI。国际多中心随机对照研究显示伊马替尼治疗的CML慢性期患者8年PFS率及OS率分别达92%和85%,远远超过传统羟基脲、干扰素等传统治疗药物的疗效[19]。
靶向VEGFR等通路治疗肝细胞癌索拉非尼可特异性作用于VEGFR,抑制其磷酸酶的活性以抑制肿瘤细胞的血管形成,并且可以通过抑制Ras-Raf-MAPK 信号通路上的 Raf 激酶进而直接抑制肿瘤细胞分裂增殖,起到抑制血管生成和抗肿瘤细胞增殖的双重作用。分别针对东西方人群的两项Ⅲ期临床研究 (SHARP 研究和Oriental 研究)已经证明索拉非尼能延长晚期HCC 患者的OS近3个月,并且可掌控不良反应,患者耐受性良好[20-21]。索拉非尼也成为目前FDA批准上市的唯一的肝细胞癌靶向治疗药物。
靶向蛋白酶体治疗NSCLC蛋白酶体是一种高度选择性的酶复合体,它能够快速降解细胞不需要的或受到损伤的靶蛋白,因为肿瘤细胞较快的增殖速度和具有缺陷的细胞周期调控,如果肿瘤细胞的蛋白质降解过程被打断,将会对肿瘤细胞增殖及存活产生巨大影响。硼替佐米是第一个应用于临床研究的蛋白酶体抑制剂,可以特异性结合20S蛋白酶体亚基发挥其抑制作用,硼替佐米治疗多发性骨髓瘤患者的APEX Ⅲ期临床试验表明治疗后患者的完全缓解率高达43%[22]。此外,新一代的蛋白酶体抑制剂卡非佐米也于2012经FDA批准上市,并且一项II期临床实验表明经治疗后的多发性骨髓瘤患者的ORR高达42%~52%[23]。
靶向治疗耐药机制
药物转运增强肿瘤细胞对药物的摄取减少、机体对药物的代谢能力增强、药物转运泵的表达或者功能发生改变与肿瘤的多药耐药密切相关。其中,ABC跨膜转运蛋白是一类最常见的药物转运泵,能够将肿瘤治疗药物转运到细胞外,从而参与肿瘤细胞的多药耐药。ABC转运蛋白超家族在所有的生命体中均有表达,在人类表达的有48种,其中的3种即ABCB 1 (P-gp/MDR1)、ABCC 1 (MRP1 /ABCC1)和 ABCG2 (BCRP/MXR)在肿瘤细胞的多药耐药中起着最重要的作用[24]。
在实体肿瘤如结肠癌、肾癌和肝癌中ABCB1高表达,高表达的ABCB1可以将底物药物泵出胞外,阻止药物发挥疗效[25]。近期研究表明靶向治疗药物如伊马替尼、厄洛替尼及苏尼替尼等可被ABCB1及ABCG2结合并排出胞外从而导致靶向治疗耐药[26]。具有原发性耐药特性的肿瘤干细胞同样也具有MDR蛋白的高表达。CD44是肿瘤干细胞的标志物之一,它的表达与患者的预后呈明显的负相关,并且CD44阳性的肿瘤细胞中往往伴有高表达的MDR蛋白,尤其是ABCG2蛋白[27]。
靶基因的改变肿瘤治疗靶点的基因突变或表达水平改变与靶向治疗耐药密切相关。EGFR-TKI类药物虽然在NSCLC的治疗中取得了令人鼓舞的疗效,但是即使是前期对EGFR-TKI治疗敏感的患者,治疗1年内也会有大约50%的患者出现耐药,其中EGFRT790M基因的二次突变是EGFR抑制剂最重要的耐药原因[28]。2015年Thress等通过检测15例应用新一代的靶向EGFRT790M基因突变的EGFR-TKI药物AZD9291后的NSCLC患者的组织后发现,获得性EGFRC797S基因突变可能是AZD9291产生耐药的机制之一[29]。ALK基因首先在间变性大细胞淋巴瘤的一个亚型中被发现,被认为是其驱动基因,而在NSCLC的患者中约有4%呈现ALK融合基因阳性,ALK基因重排对于NSCLC 靶向治疗有重大意义。克唑替尼为第一个ALK 重排抑制剂,可以通过抑制ALK 表达从而发挥作用,但大约在1年之内大多数患者不可避免的产生耐药。Choi等[30]研究表明ALK基因C1156Y及L1196M的二次突变与克唑替尼二次耐药密切相关。
靶基因旁路激活肿瘤靶基因旁路激活可以代偿因为靶基因受抑而导致的下游通路改变,从而使靶向治疗耐药。McDermott等[31]在EGFR-TKI耐药的肺癌细胞中发现c-Met 旁路扩增。Engelman等[32]也指出c-Met 旁路可以通过ErbB3并进一步激活AKT通路产生耐药。此外,虽然维罗非尼治疗后的患者PFS维持在5~6个月之间,但是一旦出现耐药,疾病呈爆发性进展,后续几乎很难有药物能控制住。目前研究表明通过COT 激活MEK[33]以及通过PDGFR β代偿MEK下游信号通路的旁路激活途径是导致维罗非尼耐药的重要原因[34]。
药物适应性上皮-间质转化上皮-间质转化 (epithelial-mesenchymal transitions,EMT)是上皮细胞在生理或病理情况下向间质细胞表型转化的现象,存在于创伤愈合、器官纤维化和肿瘤发展等过程中。Sequist等[35]在NSCLC耐药的临床标本中发现癌细胞存在明显的EMT。此外,Huang等[36]通过大范围的siRNA筛查发现MED12蛋白可以预测ALK及EGFR抑制剂的敏感性。MED12缺失可以通过激活TGF-β受体信号通路诱导NSCLC细胞发生EMT,从而导致ALK及EGFR靶向治疗耐药,而抑制TGF-β受体信号通路可以逆转因MED12缺失导致的ALK及EGFR靶向治疗耐药。另有研究通过对大量的肺癌细胞系进行EMT相关的基因筛查,将肺癌细胞系分为上皮表型或间质表型后发现肿瘤细胞的上皮或间质状态可以作为预测EGFR-TKI厄洛替尼和PI3K-AKT-mTOR信号通路抑制剂疗效的敏感性指标[37]。
肿瘤微环境改变肿瘤微环境一般是指肿瘤所处的局部生物环境,包括胞外基质、肿瘤相关成纤维细胞、免疫及炎症细胞等。肿瘤微环境与肿瘤耐药之间存在重要的病理学关联,肿瘤耐药性的机制不仅包括癌细胞内源性的改变,同时也包括肿瘤所处微环境所赋予的改变。肿瘤相关的间质细胞可以通过分泌肝细胞生长因子 (HGF)及成纤维细胞生长因子 (FGF)等激活PI3K-AKT及MEK-ERK信号通路介导耐药[38]。Straussman等[39]研究表明基质细胞分泌的HGF 可以通过增加其同源受体c-MET 的磷酸化,使BRAF-V600E基因突变细胞系抵抗BRAF 抑制剂的治疗。同样,提高肿瘤相关成纤维细胞PDGF-C 的表达能够增强抗血管生成治疗的耐药性[40]。
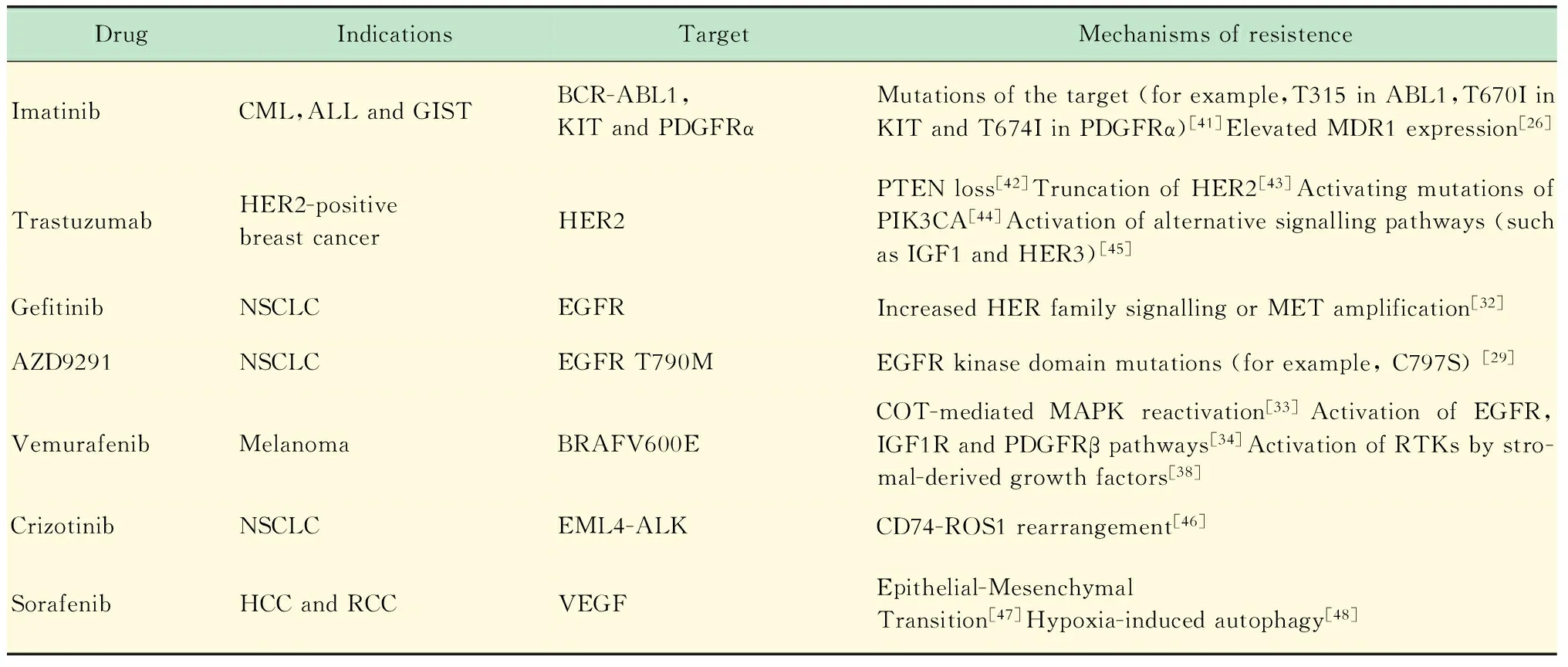
表1 重要分子靶向药物的耐药机制汇总
分子靶向药物联合用药策略
靶向药物与细胞毒性药物联合用药分子靶向药物与细胞毒性药物联合应用日益受到关注,两种不同作用机制的治疗方式联合应用可充分发挥各自优势,减少药物剂量、降低毒性、克服细胞毒类药物对肿瘤细胞内某些信号通路的激活等。Hurwitz 等[49]应用VEGFR单抗贝伐单抗联合IFL (伊立替康联合氟尿嘧啶及亚叶酸钙) 化疗方案治疗402 例转移性结肠癌的患者,结果显示,贝伐单抗联用组可明显提高患者的OS。
靶向药物联合应用不同的分子靶向药物联合应用可达到阻断信号转导通路上多个靶点和防止肿瘤耐药的目的,并且取得了较好的临床效果。靶向药物联合应用主要包括:同一靶点单抗药物与TKI药物合用、胞膜受体靶向药物与下游通路靶向药物合用及不同靶点靶向药物合用。2012年的一项Ⅲ期临床试验表明抗HER2单克隆抗体曲妥珠单抗联合HER2-TKI拉帕替尼治疗早期HER2+乳腺癌效果优于单药,此即为同一靶点单抗药物与TKI药物合用[50]。不可逆EGFR-TKI HKI-272与PI3K/Akt/mTOR通路抑制剂西罗莫司联用,能显著抑制EGFRT790M-L858R基因突变的肺癌移植瘤,此为靶向通路上下游的药物联用[51]。NSCLC的MET旁路激活为EGFR-TKI耐药的机制之一,2013年一项Ⅱ期临床试验表明联合MET靶向药物 Onartuzumab可以逆转EGFR+NSCLC癌的厄洛替尼耐药,延长患者的OS[52]。
问题及展望过去十几年,分子靶向治疗药物发展迅速,并且已经在临床的抗肿瘤治疗中发挥了极为重要的作用,而且随着分子靶向治疗药物研究的深入,将有越来越多的靶向药物进入临床。但是恶性肿瘤的发生是一个多基因、多步骤的复杂过程,并且随着肿瘤的进展可能产生出新的基因突变,增加了靶向治疗的难度。为此,研发多靶点的药物及不同靶点药物的联合应用将尤为迫切。随着对耐药机制进一步的了解以及分子生物学的发展,分子靶向治疗必将推动抗肿瘤治疗的发展,跨入一个全新的时代。
参考文献
[1]EDER J,SEDRANI R,WIESMANN C.The discovery of first-in-class drugs:origins and evolution[J].NatRevDrugDiscov,2014,13 (8):577-587.
[2]DOBBELSTEIN M,OLL U.Targeting tumour-supportive cellular machineries in anticancer drug development[J].NatRevDrugDiscov,2014,13 (3):179-196.
[3]TANDON AK,CLARK GM,CHAMNESS GC,etal.HER-2/neu oncogene protein and prognosis in breast cancer[J].JClinOncol,1989,7 (8):1120-1128.
[4]MARTY M,COGNETTI F,MARANINCHI D,etal.Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment:the M77001 study group[J].JClinOncol,2005,23 (19):4265-4274.
[5]SLAMON DJ,LEYLAND-JONES B,SHAK S,etal.Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2[J].NEnglJMed,2001,344 (11):783-792.
[6]RONOND EH,PEREZ EA,BRYANT J,etal.Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer[J].NEnglJMed,2005,353 (16):1673-1684.
[7]PICCART-GEBHART M J,PROCTER M,LEYLAND-JONES B,etal.Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer[J].NEnglJMed,2005,353 (16):1659-1672.
[8]HEINRICH MC,CORLESS CL,DUENSING A,etal.PDGFRA activating mutations in gastrointestinal stromal tumors[J].Science,2003,299 (5607):708-710.
[9]VERWEIJ J,CASALI PG,ZALCBERGJ,etal.Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib:randomised trial[J].Lancet,2004,364 (9440):1127-1134.
[10]BLANKE CD,RANKIN C,DEMETRI GD,etal.Phase Ⅲ randomized,intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase:S0033[J].JClinOncol,2008,26 (4):626-632.
[11]REICHARDT P,BLAY JY,GELDERBLOM H,etal.Phase Ⅲ study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib[J].AnnOncol,2012,23 (7):1680-1687.
[12]BLAY JY,SHEN L,KANG YK,etal.Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1):a randomised phase 3 trial[J].LancetOncol,2015,16 (5):550-560.
[13]DAVIES H,BIGNELL GR,COX C,etal.Mutations of the BRAF gene in human cancer[J].Nature,2002,417 (6892):949-954.
[14]CHAPMAN PB,HAUSCHILD A,ROBERT C,etal.Improved survival with vemurafenib in melanoma with BRAF V600E mutation[J].NEnglJMed,2011,364 (26):2507-2516.
[15]LYNCH TJ,BELL DW,SORDELLA R,etal.Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib[J].NEnglJMed,2004,350 (21):2129-2139.
[16]SEQUIST LV,MARTINS RG,SPIGEL D,etal.First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations[J].JClinOncol,2008,26 (15):2442-2449.
[17]ZHANG L,MA S,SONG X,etal.Gefitinib versus placebo as maintenance therapy in patients with locally advanced or metastatic non-small-cell lung cancer (INFORM;C-TONG 0804):a multicentre,double-blind randomised phase 3 trial[J].LancetOncol,2012,13 (5):466-475.
[18]JANNE PA,YANG JC,KIM D W,etal.AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer[J].NEnglJMed,2015,372 (18):1689-1699.
[19]DRUKER BJ,GUILHOT F,O′BRIEN SG,etal.Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia[J].NEnglJMed,2006,355 (23):2408-2417.
[20]LLOVET JM,RICCI S,MAZZAFERRO V,etal.Sorafenib in advanced hepatocellular carcinoma[J].NEnglJMed,2008,359 (4):378-390.
[21]CHENG AL,KANG YK,CHEN Z,etal.Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma:a phase Ⅲ randomised,double-blind,placebo-controlled trial[J].LancetOncol,2009,10 (1):25-34.
[22]RICHARDSON PG,SONNEVELD P,SCHUSTER M,etal.Extended follow-up of a phase 3 trial in relapsed multiple myeloma:final time-to-event results of the APEX trial[J].Blood,2007,110 (10):3557-3560.
[23]VIJ R,WANG M,KAUFMAN J L,etal.An open-label,single-arm,phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma[J].Blood,2012,119 (24):5661-5670.
[24]GOTTESMAN MM,FOJO T,BATES SE.Multidrug resistance in cancer:role of ATP-dependent transporters[J].NatRevCancer,2002,2 (1):48-58.
[25]THOMAS H,COLEY HM.Overcoming multidrug resistance in cancer:an update on the clinical strategy of inhibiting p-glycoprotein[J].CancerControl,2003,10 (2):159-165.
[26]SHUKLAS,CHEN ZS,AMBUDKAR SV.Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance[J].DrugResistUpdat,2012,15 (1-2):70-80.
[27]BHATAVDEKAR JM,PATEL DD,CHIKHLIKAR PR,etal.Overexpression of CD44:a useful independent predictor of prognosis in patients with colorectal carcinomas[J].AnnSurgOncol,1998,5 (6):495-501.
[28]SHIH JY,GOW CH,YANG PC.EGFR mutation conferring primary resistance to gefitinib in non-small-cell lung cancer[J].NEnglJMed,2005,353 (2):207-208.
[29]THRESS KS,PAWELETZ CP,FELIP E,etal.Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M[J].NatMed,2015,21 (6):560-562.
[30]CHOI YL,SODA M,YAMASHITA Y,etal.EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors[J].NEnglJMed,2010,363 (18):1734-1739.
[31]MCDERMOTT U,SETTLEMAN J.Personalized cancer therapy with selective kinase inhibitors:an emerging paradigm in medical oncology[J].JClinOncol,2009,27 (33):5650-5659.
[32]ENGELMAN JA,ZEJNULLAHU K,MITSUDOMI T,etal.MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling[J].Science,2007,316 (5827):1039-1043.
[33]JOHANNESSEN CM,BOEHM JS,KIM SY,etal.COT drives resistance to RAF inhibition through MAP kinase pathway reactivation[J].Nature,2010,468 (7326):968-972.
[34]NAZARIAN R,SHI H,WANG Q,etal.Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation[J].Nature,2010,468 (7326):973-977.
[35]SEQUIST LV,WALTMAN BA,DIAS-SANTAGATA D,etal.Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors[J].SciTranslMed,2011,3 (75):26r-75r.
[36]HUANG S,HOLZEL M,KNIJNENBURG T,etal.MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling[J].Cell,2012,151 (5):937-950.
[37]BYERS LA,DIAO L,WANG J,etal.An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance[J].ClinCancerRes,2013,19 (1):279-290.
[38]WILSON TR,FRIDLYAND J,YAN Y,etal.Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors[J].Nature,2012,487 (7408):505-509.
[39]STRAUSSMAN R,MORIKAWA T,SHEE K,etal.Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion[J].Nature,2012,487 (7408):500-504.
[40]CRAWFORD Y,KASMAN I,YU L,etal.PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment[J].CancerCell,2009,15 (1):21-34.
[41]CHEN Y,TAKITA J,CHOI YL,etal.Oncogenic mutations of ALK kinase in neuroblastoma[J].Nature,2008,455(7215):971-974.
[42]NAGATA Y,LAN K H,ZHOU X,etal.PTEN activation contributes to tumor inhibition by trastuzumab,and loss of PTEN predicts trastuzumab resistance in patients[J].CancerCell,2004,6 (2):117-127.
[43]RECUPERO D,DANIELE L,MARCHIO C,etal.Spontaneous and pronase-induced HER2 truncation increases the trastuzumab binding capacity of breast cancer tissues and cell lines[J].JPathol,2013,229 (3):390-399.
[44]BERNS K,HORLINGS HM,HENNESSY BT,etal.A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer[J].CancerCell,2007,12 (4):395-402.
[45]LU Y,ZI X,ZHAO Y,etal.Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin)[J].JNatlCancerInst,2001,93 (24):1852-1857.
[46]BERGETHON K,SHAW AT,OU SH,etal.ROS1 rearrangements define a unique molecular class of lung cancers[J].JClinOncol,2012,30 (8):863-870.
[47]ZHANG W,SUN HC,WANG WQ,etal.Sorafenib down-regulates expression of HTATIP2 to promote invasiveness and metastasis of orthotopic hepatocellular carcinoma tumors in mice[J].Gastroenterology,2012,143 (6):1641-1649.
[48]PAEZ-RIBES M,ALLEN E,HUDOCK J,etal.Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis[J].CancerCell,2009,15 (3):220-231.
[49]HURWITZ H,FEHRENBACHER L,NOVOTNY W,etal.Bevacizumab plus irinotecan,fluorouracil,and leucovorin for metastatic colorectal cancer[J].NEnglJMed,2004,350 (23):2335-2342.
[50]BASELGA J,BRADBURY I,EIDTMANN H,etal.Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO):a randomised,open-label,multicentre,phase 3 trial[J].Lancet,2012,379 (9816):633-640.
[51]ENGELAMAN JA,CHEN L,TAN X,etal.Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers[J].NatMed,2008,14 (12):1351-1356.
[52]SPIGEL DR,ERVIN T J,RAMLAU RA,etal.Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer[J].JClinOncol,2013,31 (32):4105-4114.
Research progresses in molecular targeted therapy of cancer
ZHANG Yu1,2, KADEL Dhruba1,2, SUN Hao-ran1,2, QIN Lun-xiu1,2,3△
(1DepartmentofGeneralSurgery,HuashanHospital,FudanUniversity,Shanghai200040,China;2CancerMetastasisInstitute,FudanUniversity,Shanghai200040,China;3InstituteofBiomedicalSciences,FudanUniversity,Shanghai200032,China)
【Abstract】In recent decade,a number of targeted therapies have been discovered and proven effective in a variety of human hematological and solid malignancies.However,the relatively rapid acquisition of resistance to such treatments which is observed in virtually all cases significantly limits their utility and remains a substantial challenge to their clinical application.As molecular mechanisms of resistance have begun to be elucidated,new strategies to overcome or prevent the development of resistance have been emerging.Here,we summarize the characteristics of these targeted therapies and provide an overview of the key clinical trials that led to approval of these drugs,the various mechanisms of drug resistance and potential ways to overcome that.
【Key words】cancer;targeted therapy;drug resistance;combination therapy
【中图分类号】R730.53
【文献标识码】B
doi:10.3969/j.issn.1672-8467.2016.01.021
(收稿日期:2015-11-10;编辑:沈玲)
△Corresponding authorE-mail:qinlx@fudan.edu.cn
猜你喜欢
保健医苑(2022年5期)2022-06-10
现代临床医学(2022年3期)2022-06-06
中国典型病例大全(2022年7期)2022-04-22
昆明医科大学学报(2022年1期)2022-02-28
中国医药导报(2016年29期)2016-12-27
中国实用医药(2016年29期)2016-12-26
上海医药(2016年23期)2016-12-22
上海医药(2016年22期)2016-12-13
医学信息(2016年29期)2016-11-28
中国实用医药(2016年26期)2016-11-07
