
17β-雌二醇和2-甲氧基雌二醇对慢性低氧性肺动脉高压大鼠体内内皮素-1/一氧化氮体系的影响
2016-06-13 02:29王丽汪佩李岩鹏袁雅冬
中国循环杂志 2016年5期
王丽,汪佩,李岩鹏,袁雅冬
17β-雌二醇和2-甲氧基雌二醇对慢性低氧性肺动脉高压大鼠体内内皮素-1/一氧化氮体系的影响
王丽,汪佩,李岩鹏,袁雅冬
摘要
目的:探讨17β-雌二醇(E2)及2-甲氧基雌二醇(2ME)对慢性低氧性肺动脉高压大鼠体内内皮素-1(ET-1)/一氧化氮(NO)体系的影响。
方法:雌性SD大鼠48只,按随机数字表法平均分为6组(各组n=8):假手术组、去势组、低氧组、去势+低氧组、去势+低氧+E2组、去势+低氧+2ME组。去势组行卵巢切除术;非去势组打开腹腔找到卵巢后不做处理直接还纳缝合。术后去势+低氧+E2组皮下注射E2[20 μg/(kg·d)],去势+低氧+2ME组皮下注射2ME[240 μg/(kg·d)],其他组皮下注射生理盐水(0.1 ml/d)。低氧组大鼠置于低氧箱内饲养,非低氧组大鼠呼吸正常空气。连续饲养8周以建立低氧性肺动脉高压模型。分别测定血清ET-1、NO水平、内皮型一氧化氮合酶(eNOS)活性及肺组织内皮素A受体(ETAR)、内皮素B受体(ETBR)、eNOS 表达变化。
结果:低氧组、去势+低氧组大鼠肺小动脉管壁增厚,管腔变窄明显,平均肺动脉压(mPAP)显著高于假手术组(P均<0.01);E2和2ME干预组大鼠上述形态学及mPAP改变相对较轻。与假手术组相比,去势组和低氧组血清ET-1和肺组织ETAR mRNA及蛋白水平均明显升高,ETBR mRNA及蛋白水平明显下降(P均<0.01);去势+低氧组以上指标变化更显著;E2和2ME干预后血清ET-1和肺组织ETAR表达明显下降,但仍高于假手术组,同时ETBR 表达明显上升,但仍低于假手术组(P均<0.01);且去势+低氧+2ME组血清ET-1水平低于去势+低氧+E2组(P<0.05)。与假手术组相比,去势组肺组织eNOS蛋白水平明显下降(P<0.01);低氧组血清NO水平及肺组织eNOS蛋白水平明显下降(P<0.05或P<0.01);去势+低氧组血清NO水平、eNOS活性及肺组织eNOS mRNA和蛋白水平均明显下降(P均<0.01);去势+低氧+E2组和去势+低氧+2ME组上述指标较去势+低氧组均明显上升(P<0.05或P<0.01),但仍低于假手术组(P均<0.05)。
结论:E2及2ME均能显著降低血清ET-1水平,减少肺组织ETAR表达,增加ETBR的表达,升高血清NO水平、eNOS活性及肺组织eNOS表达,通过改善ET-1/NO体系平衡部分逆转肺动脉高压。
关键词高血压,肺性;一氧化氮;17β-雌二醇;2-甲氧基雌二醇;内皮素-1
作者单位:050000河北省石家庄市,河北医科大学第二医院呼吸与危重症医学科
(Chinese Circulation Journal,2016,31:489.)
低氧性肺动脉高压(HPH)是临床多种心肺疾病发生发展过程中重要的病理生理环节,其形成机制尚不完全清楚。急性缺氧可导致肺动脉平滑肌细胞可逆性的收缩,是一种保护性的生理反应。而慢性缺氧会使肺血管发生不可逆性的重塑,是形成和加重肺动脉高压的重要原因。HPH的病理生理基础包括肺血管收缩、结构重建、微血栓形成,其发生发展涉及多种血管活性物质,其中内皮功能障碍起关键性作用,缺氧致血管内皮受损、功能失调,引起血管活性物质和细胞因子产生及释放异常,血管舒缩因子平衡失调,包括内皮素-1(ET-1)、一氧化氮(NO)等,最终导致肺动脉高压发生[1]。
近年来,关于雌激素及其代谢产物对肺血管起保护作用的研究日益增多,因其可减弱多种刺激引起的肺血管内皮细胞和平滑肌细胞功能损伤,有利于改善血管重构和舒张血管,但具体机制目前尚未完全了解。本研究旨在通过建立HPH动物模型探讨HPH时ET-1/NO体系的变化情况和二者之间的相互关系以及17β-雌二醇(E2)、2-甲氧基雌二醇(2ME)是否通过影响ET-1/NO体系平衡而在肺动脉高压中起保护作用。
1 材料与方法
主要仪器与试剂:常压低氧环境动物实验箱(长沙长锦科技有限公司);RM3000四导联生理记录仪(日本光电公司);Eclipse 55i光学显微镜及照相系统(日本尼康);高速冷冻离心机1-15K (美国Sigma公司);离心机(上海安亭);756MC型紫外-可见分光光度计(上海精密科学仪器有限公司);E2、2ME(进口分析纯度,美国Sigma公司);125I-ET放射免疫分析试剂盒(北京北方生物研究所);NO试剂盒、NOS试剂盒(南京建成生物工程研究所);PCR Reagents、总RNA提取试剂(大连TaKaRa公司);大鼠抗肺组织内皮素A受体(ETAR)单克隆抗体、大鼠抗内皮素B受体(ETBR)单克隆抗体、辣根过氧化物酶标记的二抗(美国 Santa Cruz公司)。
实验动物及分组:6~8周龄健康雌性SD大鼠 48只,体重(180±10)g,由河北医科大学实验动物中心提供。按随机数字表法将大鼠平分为6组:假手术组、去势组、低氧组、去势+低氧组、去势+低氧+E2组、去势+低氧+2ME组(各组n=8)。去势组大鼠打开腹腔,沿输卵管找到卵巢并结扎切除;非去势组找到卵巢后不做处理直接还纳缝合。术后假手术组、去势组、低氧组、去势+低氧组皮下注射生理盐水(0.1 ml/d),去势+低氧+E2组皮下注射E2[20μg/(kg·d)],去势+低氧+2ME组皮下注射2ME[240μg/(kg·d)]。待伤口愈合后,低氧组大鼠置于低氧箱内,保持氧浓度为(10.0±0.5)%,以钠石灰和无水氯化钙吸收多余的二氧化碳(CO2)和水蒸气,控制CO2浓度低于0.5%,每天持续低氧24 h,连续8周。非低氧组大鼠置于同一室内饲养,除吸入空气外,其它条件与低氧组相同。
肺动脉压力测定及肺小动脉形态学分析:大鼠经1%戊巴比妥钠(40 mg/kg)麻醉后,钝性分离右侧颈外静脉,自右颈外静脉插入微导管,将压力传感器接上生理记录仪进行连续测压,记录肺动脉压力波形,计算平均肺动脉压(mPAP)。
肺动脉压力检测后,留取血标本2 ml,3000 rpm离心10 min,取上清液迅速放入-80℃冰箱保存,待测ET-1、NO含量和内皮型一氧化氮合酶(eNOS)活性。处死大鼠,快速打开胸腔取出全肺,以生理盐水冲洗肺外表,取左肺上叶制成石蜡标本,行苏木素-伊红(HE)染色,镜下观察肺动脉形态;余肺叶待测ETAR、ETBR及eNOS mRNA和蛋白水平。
血清ET-1、NO水平及eNOS活性测定:应用放射免疫法测定血清ET-1浓度[2],按125I-ET放射免疫分析试剂盒检测;采用硝酸还原酶法测定血清NO浓度[2],按NO试剂盒说明书进行检测。应用化学法测定血清eNOS活性[3],按NOS试剂盒说明书进行检测。
肺组织ETAR、ETBR及eNOS mRNA的测定:逆转录多聚酶链反应(RT-PCR)法检测肺组织ETAR、ETBR 及eNOS mRNA表达:用Trizol法提取组织总RNA,取2 µg总RNA逆转录成cDNA。引物分别为:ETAR 退火温度为 55℃,上游引物5'-CGTCCTGGGAAGTTTCCTCC-3 ',下游引物 5'-ATTGCTAGGCAGGGCCAAAT-3',产物长度为371bp;ETBR 退火温度为56℃,上游引物5'-CTAAGGCAGTTGAGGACCTGG-3',下游引物 5'-GTCTTAGTGGGTGGCGTCAT-3',产物长度为282 bp;eNOS 退火温度为56℃,上游引物 5'-CGAACAGCAGGAGCTAGAGG-3'、下游引物 5'-GAGGTGGATCTCTCCTGGGT-3 ',产物长度为211 bp;甘油醛-3-磷酸脱氢酶(GAPDH)上游引物 5'-CACCTTTGATGCTGGGGCTG-3 ',下游引物 5'-TGGTATTCGAGAGAAGGGAGGG-3 ',产物长度为306 bp。反应体系含Taq DNA聚合酶,dNTPs,上样染料,稳定剂,优化剂,反应缓冲液,逆转录反应产物,上下游引物等。反应程序为94 ℃预变性4 min,然后进入PCR循环,94 ℃变性30 s,各自退火温度30 s,72 ℃延伸1 min,循环35次后再延伸7 min。PCR产物在1.5%琼脂糖凝胶上电泳后,应用复日FR-980A生物凝胶电泳图像分析系统对PCR产物的电泳带进行紫外光密度扫描成像,应用图像分析软件对目的条带进行分析,以内参条带作为参照,结果以两者的吸光度比值表示。
肺组织ETAR、ETBR 及eNOS 蛋白的测定:蛋白免疫印迹法(Western blot)检测肺组织ETAR、ETBR 及eNOS蛋白表达:肺组织总蛋白的提取采用RIPA裂解液,蛋白含量按改良Lowry法测定[4]。取总蛋白60 μg经15%SDS-聚丙烯酰胺凝胶电泳后,转移至聚偏二氟乙烯膜(PVDF)上。转膜完毕,取出 PVDF膜,置于含 5 % 脱脂奶粉的 TTBS( 10 mmol/L Tris-HCl,pH 8.0,150 mmol/L NaCl,0.05 % Tween-20)封闭液中,室温2 h。将封闭后的膜置入TTBS适当稀释的一抗溶液中,4℃过夜。洗膜后将膜置入适量以TTBS 1:10000 稀释的辣根过氧化物酶标记的二抗溶液中,室温反应 2.5~3 h。用TTBS洗膜3次。最后扫描成像并用 Vilber Lourmat公司ID 数码成像分析软件对Western blot显色区带的信号强度进行相对定量分析,扫描灰度值用积分光密度值表示。
统计学方法:采用 SPSS13.0 统计软件进行处理,正态分布的计量数据以±s表示,多个样本均数两两之间比较用方差分析中的LSD和SNK-q 检验,以 P<0.05为差异有统计学意义。
2 结果
2.1 成功建立肺动脉高压模型(图1、2)
假手术组及去势组大鼠反应敏捷,体毛光泽,饮食正常;低氧条件下的各组大鼠随着低氧时间的延长,反应迟钝变得虚弱,毛色晦暗,呼吸急促,食水量和活动减少,最终以静卧为主。

图1 各组大鼠肺组织形态学变化(苏木素伊红,×100)
光镜下,假手术组及去势组肺动脉内膜完整平滑,管壁菲薄,肌层无增厚;低氧 8周后,可见肺动脉管壁和平滑肌层明显增厚,管腔狭窄,表现出肺动脉重构的体征,mPAP明显升高,去势+低氧组上述形态学及mPAP改变最显著,E2和2ME干预组上述改变相对较轻。
2.2 血清ET-1水平及肺组织ETAR、ETBR mRNA和蛋白表达的变化(图3~5)
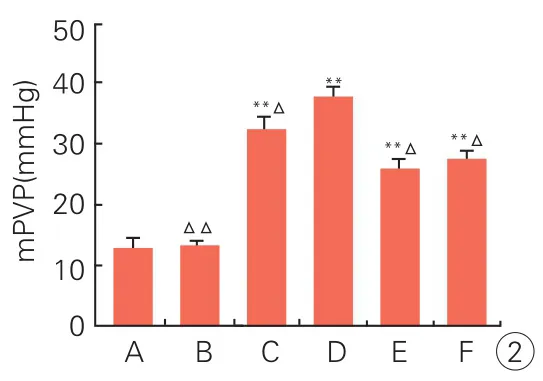
图2 各组大鼠平均肺动脉压(mPAP)变化(±s,各组n=8)

图3A-3C 各组大鼠血清ET-1和NO水平及eNOS活性的比较(±s,各组n=8)

图4 各组大鼠肺组织中ETAR、ETBR、eNOS mRNA表达的逆转录多聚酶链反应电泳图及其水平的比较(±s,各组n=8)

图5 各组大鼠肺组织中ETAR、ETBR、eNOS 的蛋白免疫印迹分析的印迹图及其蛋白水平比较 (±s,各组n=8)
与假手术组相比,去势组和低氧组血清ET-1水平和肺组织ETAR mRNA及蛋白表达水平均明显升高,同时肺组织ETBR mRNA和蛋白表达水平明显下降(P均<0.01);去势+低氧组上述指标变化更显著;E2和2ME干预后血清ET-1和肺组织ETAR表达明显下降,但仍高于假手术组水平,ETBR 表达明显升高,但仍低于假手术组水平(P均<0.01),且去势+低氧+2ME组血清ET-1水平低于去势+低氧+E2组(P<0.05)。
2.3 血清NO水平、eNOS活性及肺组织eNOS mRNA和蛋白表达的变化(图3~5)
与假手术组相比,去势组肺组织eNOS蛋白水平明显下降(P<0.01);低氧组血清NO水平及肺组织eNOS蛋白水平均明显下降(P<0.05或P<0.01);去势+低氧组血清NO水平、eNOS活性及肺组织eNOS mRNA和蛋白表达水平均显著下降(P均<0.01)。去势+低氧+E2组和去势+低氧+2ME组上述指标较去势+低氧组均明显上升(P<0.05或P<0.01),但仍低于假手术组(P均<0.05)。
3 讨论
HPH是由低氧引起血管内皮细胞损伤,血管内皮合成和分泌的各种血管舒缩因子平衡失调,导致早期的肺血管收缩以及后期的肺血管重建[5]。在本实验中以雌性大鼠作为研究对象,低氧8周后mPAP明显上升,肺小动脉管壁增厚,成功地建立肺动脉高压模型。在低氧诱导的肺动脉高压模型中,大鼠血清ET-1水平、肺组织ETAR 表达明显增加,ETBR 表达明显减少;血清NO水平、eNOS活性及肺组织eNOS表达均明显下降。
ET-1/NO体系平衡失调在肺动脉高压发生发展过程中发挥着重要作用,但二者之间具体始动关系尚不明确。ET-1是迄今己知的作用最强大的内源性缩血管肽,主要通过ETAR、ETBR两种受体发挥作用。低氧条件下,ETAR激活后使Ca2+内流增加,同时激活磷酯酶A2和C、蛋白激酶C,并能增强c-myc、c-fos、c-jun等一系列与细胞增殖密切相关的原癌基因的表达[6,7],最终引起肺血管收缩和结构重塑。而血管内皮细胞ETBR增加可以促进NO及前列环素(PGI2)释放[8],减少内皮素转化酶的表达[9],同时灭活ET-1[10],从而引起肺血管舒张,对抗肺血管重塑进展。NO作为体内广泛分布的血管内皮舒张因子,通过激活血管平滑肌细胞(VSMC)中的鸟苷酸环化酶,使细胞内环磷酸鸟苷(cGMP)大量产生并堆积,从而导致血管舒张。另外,NO还能够通过抑制ET的合成、释放,调节活性因子儿茶酚胺等所致的血管收缩这一途径调节血管舒缩平衡,在一定程度上能够逆转低氧性肺血管收缩。通常认为肺动脉高压的病理改变同NO生物活性降低和(或)NO合成减少有关[11]。eNOS 是血管内皮细胞生成NO的限速酶[12],它在调节肺血管张力中起着非常重要的作用[13]。本研究再次证实了ET-1/NO体系失衡参与了HPH的发生和发展过程。并且ET-1体系变化较NO体系变化更显著,说明ET-1体系可能起始动作用,其作用增强,抑制NO体系表达,引起NO体系下调,ET-1/NO比值上调,导致肺动脉压力升高,肺动脉平滑肌增生,血管重构。
雌激素在肺血管系统起保护作用的研究日趋增多,但具体机制仍不是很明确。有研究报道,在慢性HPH大鼠模型中,去卵巢大鼠发生肺动脉重构和右心室肥大的程度更加明显[14,15]。补充雌激素治疗后可降低肺动脉高压严重程度[16,17],但并未完全抑制和逆转,这可能与常压明显缺氧时引起内皮细胞的损伤较严重有关。Yuan等[18]认为E2与雌激素受体β结合激活磷脂酰肌醇3激酶(PI3K)/蛋白激酶B(Akt)通路来降低ET-1的表达从而改善肺动脉高压。Earley等[19]认为E2主要通过干扰低氧诱导因子活性以及雌激素受体与低氧诱导因子通路竞争限制cAMP反应元件结合蛋白结合蛋白(CBP)/p300转录共激活因子的数量从而抑制ET-1的表达。另外,有研究认为E2与雌激素受体结合通过转录调控激活eNOS基因的表达,使eNOS活性或蛋白表达增加[20],进而促进NO释放。也有研究报道,E2可与雌激素膜受体结合增加细胞内Ca2+浓度,进而使eNOS活性增加,从而产生更多的NO[21]。本研究发现E2干预后大鼠血清ET-1水平、肺组织ETAR表达均明显减少,而ETBR表达增加;并且肺组织eNOS表达、血清eNOS活性及NO水平均升高。这说明E2可有效对抗低氧引起的ET-1/NO体系失衡从而抑制HPH发生。分析其原因可能为,E2抑制肺血管内皮细胞合成和分泌ET-1,并减少肺动脉平滑肌细胞上ETAR的表达,减弱了ET-1与ETAR的作用及其介导的NO及NOS表达的下降[22,23],同时增强了血管内皮细胞上的ETBR清除ET-1并介导释放舒血管因子(如NO和PGI2)[24]的作用,抑制了ET-1引起的肺血管收缩和重构,并增强了血管的舒张功能。总之,E2抑制HPH,可能是通过抑制ET-1途径,增加 eNOS的活性和NO 的含量,促进ET-1/NO 稳态的恢复来实现的。
2ME为E2的主要非雌激素性代谢产物,与雌激素受体的亲和力极低,有很少或无雌激素活性。2ME具有抗肿瘤及抗VSMC增殖作用,且能够不依赖雌激素受体而独立发挥作用[25],由于其不良反应小,极具研究价值。有研究表明,2ME能通过细胞应激反应引起线粒体细胞色素C释放、促进活性氧生成、P53表达以及活化蛋白激酶(JNK)和Bcl-2家族的活性[26],导致细胞调亡。也有研究发现,2ME可诱导细胞周期停滞在G0/G1期和G2/M期,还可抑制细胞外信号调节激酶(ERK)1/2和Akt信号通路,从而抑制人大动脉平滑肌细胞增殖[27]。本研究发现2ME可降低血清ET-1水平,抑制肺组织ETAR表达,增强ETBR表达,同时提高血清NO水平、eNOS活性及肺组织eNOS表达,改善ET-1/NO体系平衡,缓解肺动脉压力以及肺血管重塑改变;且与Dubey等[28]研究结果一致,2ME抑制ET-1的能力明显高于E2。提示,2ME抑制HPH的发生发展也与其改善ET-1/NO体系平衡作用有关,且可能具有不同于E2的作用途径,而与其诱导细胞凋亡以及抑制细胞有丝分裂是否有关,尚需进一步研究。
总之,E2及2ME均能够抑制血清ET-1水平、肺组织ETAR表达,促进ETBR表达,提高eNOS表达及活性,增加血清NO水平,使ET-1和NO体系之间的失衡状态得到改善,从而改善内皮功能,抑制肺血管收缩及结构重塑,降低肺动脉压力。
参考文献
[1]d'Uscio LV.eNOS uncoupling in pulmonary hypertension.Cardiovasc Res,2011,92:359-360.
[2]Camsarl A,Pekdemir H,Cicek D,et al.Endothelin-1 and nitric oxide concentrations and their response to exercise in patients with slow coronary flow.Circ J,2003,67:1022-1028.
[3]Mookerjee RP,Wiesenthal A,Icking A,et al.Increased gene and protein expression of the novel eNOS regulatory protein NOSTRIN and a variant in alcoholic hepatitis.Gastroenterology ,2007,132:2533-2541.
[4]Shen YX,Xiao K,Liang P,et al.Improvement on the modified Lowry method against interference of divalent cations in soluble proteinmeasurement.Appl Microbiol Biotechnol,2013 ,97:4167-4178.
[5]Nitta CH,Osmond DA,Herbert LM,et al.Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension.Am J Physiol Heart Circ Physiol,2014,306:H41-H52.
[6]Kapakos G,Bouallegue A,Daou GB,et al.Modulatory Role of Nitric Oxide/cGMP System in Endothelin-1-induced signaling responses in vascular smooth muscle cells.Curr Cardiol Rev,2010,6:247-254.
[7]Takuwa NY,Takuwa M,Yanagisawa M,et al.A novel vasoactive peptide endothelin stimulates mitogenesis through inosito lipid turnover in Swiss 3T3 fibroblast .J Biol Chem,1989,264:7856-7861.
[8]Bourque SL,Davidge ST,Adams MA.The interaction between endothelin-1 and nitric oxide in the vasculature:new perspectives.Am J Physiol Regul Integr Comp Physiol,2011,300:R1288-R1295.
[9]Chester AH,Yacoub MH.The role of endothelin-1 in pulmonary arterial hypertension.Glob Cardiol Sci Pract,2014,2014 :62-78.
[10]Mazzuca MQ,Khalil RA.Vascular endothelin receptor type B:structure,function and dysregulation in vascular disease.Biochem Pharmacol,2012,84:147-162.
[11]Coggins MP,Bloch KD.Nitric oxide in the pulmonary vasculature.Arterioscler Thromb Vasc Biol,2007,27:1877-1885.
[12]Man HSJ,Tsui AKY,Marsden PA,et al.Nitric oxide and hypoxia signaling.Nitric Oxide,2014,96:161-192.
[13]Gielis JF,Lin JY,Wingler K,et al.Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders .Chin J Integr Med.Free Radic Biol Med,2011,50:765-776.
[14]袁梦琪,段争,袁雅冬,等.雌激素对低氧性肺动脉高压ACEAngⅡ-AT1 轴的影响.中华医学杂志,2014,94:1696-1701.
[15]Resta TC,Kanagy NL,Walker BR.Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression Am J Physiol Lung Call Mol physio1,2001,280:L88-97.
[16]Pak O,Aldashev A,Welsh D,et al.The effects of hypoxia on the cells of the pulmonary vasculature.Eur Respir J,2007,30:364-372.
[17]Austin ED,Cogan JD,West JD,et al.Alterations in oestrogen metabolism:implications for higher penetrance of familial pulmonary arterial hypertension in females.Eur Respir J,2009,34:1093-1099.
[18]Yuan P,Wu WH,Gao L,et al.Oestradiol ameliorates monocrotaline pulmonary hypertension via NO,prostacyclin and endothelin-1 pathways.Eur Respir J,2013,41:1116-1125.
[19]Earley S,Resta TC.Estradiol attenuates hypoxia-induced pulmonary endothelin-1 gene expression.Am J Physiol Lung Cell Mol Physiol,2002,283:L86-L93.
[20]Hox V,Desai A,Bandara G,et al.Estrogen increases the severity of anaphylaxis in female mice through enhanced endothelial nitric oxide synthase expression and nitric oxide production.J Allergy Clin Immunol,2015,135:729-736.
[21]Reslan OM,Khalil RA.Vascular effects of estrogenic menopausal hormone therapy.Rev Recent Clin Trials,2012 ,7:47-70.
[22]Jia Y,Zhao Z,Xu M,et al.Prevention of renal ischemia-reperfusion injury by short hairpin RNA of endothelin A receptor in a rat model.Exp Biol Med (Maywood),2012 ,237:894-902.
[23]栾丽丽,李志超,黄玉芳,等.NO及NOS在低氧大鼠肺组织和主动脉中的差异性及其意义.心脏杂志,2007,19:258-261.
[24]Kelland NF,Kuc RE,McLean DL,et al.Endothelial cell-specific ETB receptor knockout:autoradiographic and histological characterisation and crucial role in the clearance of endothelin-1.Can J Physiol Pharmacol,2010,88:644-651.
[25]Lakhan NJ,Sarkar MA,Venitz J,et al.2-methoxyestradiol,a promising anticancer agent.Pharmacotherapy,2003,23:165-172.
[26]Lis A,Ciesielski MJ,Barone TA,et a1.2-Methoxyestradiol inhibits proliferation of normal and neoplastic glial cells,and induces cell death,in vitro.Cancer Lett,2004,213:57-65.
[27]Barchiesi F,Jackson EK,Fingerle J,et a1.2-Methoxyestradiol,an estradiol metabolite,inhibits neointima formation and smooth muscle cell growth via double blockade of the cell cycle.Circ Res,2006,99:266-274.
[28]Dubey RK,Jackson EK,Keller PJ,et al.Estradiol metabolites inhibit endothelin synthesis by an estrogen receptor-independent mechanism.Hypertension,2001,37:640-644.
(编辑:梅平)
Effects of 17 β-estradiol and 2-methoxyestradiol on Endothelin-1/Nitric Oxide Cascade in Experimental Rats With Hypoxic Pulmonary Hypertension
WANG Li,WANG Pei,LI Yan-peng,YUAN Ya-dong.
Department of Respiratory and Critical Care Medicine,Second Hospital,Hebei Medical University,Shijiazhuang (050000),Hebei,China
Abstract
Objective:To explore the effects of 17 β-estradiol (E2) and 2-methoxyestradiol (2ME) on endothelium-1/nitric oxide (ET-1/NO) cascade in experimental rats with hypoxic pulmonary hypertension.
Methods:A total of 48 female SD rats were randomly divided into 6 groups:①Sham operation group,②Ovariectomy (OVX) group,③Hypoxia group,④OVX+hypoxia group,⑤OVX+hypoxia+E2 group,the rats received subcutaneous E2 at 20μg/(kg•d) and ⑥OVX+hypoxia+2ME group,the rats received subcutaneous 2ME at 240μg/(kg•d).n=8 in each group.Blood levels of ET-1,NO,eNOS activity and the expressions of pulmonary tissue endothelium A receptor (ETAR),ETBR and eNOS were compared among different groups.
Results:Compared with Sham operation group,Hypoxia and OVX+hypoxia groups showed small pulmonary arterythickening with lumen narrowing,increased mean pulmonary arterial pressure (mPAP),all P<0.01; the above morphological and mPAP changes were reduced by E2 and 2ME intervention.Compared with Sham operation group,OVX and Hypoxia groups had increased blood ET-1 and pulmonary mRNA,protein expressions of ETAR,decreased pulmonary ETBR,all P<0.01; the above changes were more obvious in OVX+hypoxia group; E2 and 2ME intervention reduced blood ET-1 and pulmonary ETAR expression,but they were still higher than Sham operation group,meanwhile,ETBR expression was elevated,but it was still lower than Sham operation group,all P<0.01; blood ET-1 was lower in OVX+hypoxia+2ME group than OVX+hypoxia+E2 group,P<0.05.Compared with Sham operation group,OVX group had decreased pulmonary eNOS protein expression,P<0.01; Hypoxia group had decreased blood NO and pulmonary eNOS protein expression,P<0.05 or P<0.01; OVX+hypoxia group had decreased blood NO,eNOS activity and decreased pulmonary mRNA and protein expressions of eNOS,all P<0.01; E2 and 2ME intervention elevated the above indexes,P<0.05 or P<0.01,but they were still lower than Sham operation group,all P<0.05.
Conclusion:E2 and 2ME could decrease blood ET-1 and pulmonary ETAR expression,increase pulmonary ETBR expression; elevate blood NO,eNOS activity and pulmonary eNOS expression.E2 and 2ME may partially reverse pulmonary hypertension via improving ET-1/NO cascade in experimental rats.
Key wordsHypertension,pulmonary; Nitric oxide; 17 Beta-estradiol; 2-methoxyestradiol; Endothelin-1
基金项目:河北省自然科学基金(H2013206403)
作者简介:王丽硕士研究生主要从事呼吸系统疾病研究Email:wangliarrive@163.com通讯作者:袁雅冬Email:yyd1108@126.com
中图分类号:R541
文献标识码:A
文章编号:1000-3614(2016)05-0489-06
doi:10.3969/j.issn.1000-3614.2016.05.017
Corresponding Author:YUAN Ya-dong,Email:yyd1108@126.com
收稿日期:(2015-07-04)
猜你喜欢
猪业科学(2022年8期)2022-11-22
昆明医科大学学报(2021年6期)2021-07-31
天津医科大学学报(2021年3期)2021-07-21
现代临床医学(2021年2期)2021-03-29
心肺血管病杂志(2020年5期)2021-01-14
猪业科学(2020年11期)2020-12-17
中国特种设备安全(2019年4期)2019-05-20
国外畜牧学·猪与禽(2018年11期)2018-05-14
中国男科学杂志(2016年5期)2016-12-01
中国体外循环杂志(2015年3期)2015-12-08
