
改进HPLC法测定人血清中丙戊酸钠的浓度
2016-06-06 08:41沈陈军陈新贵胡秀萍
皖南医学院学报 2016年1期
沈陈军,陈新贵,王 勇,胡秀萍
(滁州市第一人民医院 药剂科临床药学室,安徽 滁州 239000)
改进HPLC法测定人血清中丙戊酸钠的浓度
沈陈军,陈新贵,王勇,胡秀萍
(滁州市第一人民医院药剂科临床药学室,安徽滁州239000)
【摘要】目的:建立改进HPLC法测定人血清中丙戊酸钠的浓度,为临床合理使用丙戊酸钠提供依据。方法:环己烷羧酸作为内标,血样经硫酸酸化和蛋白沉淀,用正己烷提取后,与2-溴苯乙酮和三乙胺催化衍生,使用HPLC进行测定。色谱柱:ZORBAX SB-C18柱(150 mm×4.6 mm,5 μm);流动相:甲醇-水(72∶28);流速:1.0 mL/min;紫外检测波长:248 nm;柱温:25℃;进样量:10 μL。结果:血清中丙戊酸钠线性范围13.3~160.7 μg/mL(r=0.9979,n=6),血清丙戊酸钠低、中、高浓度方法回收率和萃取回收率分别为88.6%~107.2%和82.1%~89.5%,日内精密度≤6.4%,日间精密度≤8.2%。结论:本实验改进后的方法特异性强、准确度高、线性关系良好、节约实验室成本等,可适用于临床丙戊酸钠常规血药浓度监测的需求。
【关键词】HPLC;丙戊酸钠;血药浓度;药动学
【DOI】10.3969/j.issn.1002-0217.2016.01.006
丙戊酸钠又叫二丙基乙酸钠,是临床上常用的抗癫痫药,但其副作用明显,其中较严重的是导致血液系统损害和肝肾功能的异常等。由于使用量大,在临床上常有不良反应事件的发生。丙戊酸钠有效血药浓度在50~100 μg/mL之间,由于给药剂量和其体内过程与疗效有较大差异,并且其剂量与药效的相关度明显小于血药浓度与药效的相关度,因此对丙戊酸钠进行临床常规血药浓度监测是很有必要的。目前丙戊酸钠血药浓度检测采用免疫法和HPLC法,对已发表的大量HPLC法测定丙戊酸钠血药浓度的文献进行分析和实验室验证发现,存在如下问题[4-5]:色谱条件选择差异大,应遵循色谱结果最佳且节约试剂的目的;萃取提取剂选择不一,其中乙醚挥发性大并且有毒;衍生催化剂三乙胺加入量不统一,且相应实验结果有较大差异;水浴衍生过程阐述不明,如水浴温度、时间、水浴后萃取剂挥发方法不明等。因此通过改进现有实验室条件和方法,本实验室建立了用硫酸酸化,正己烷萃取,三乙胺催化,2-溴苯乙酮衍生化和HPLC检测的实验方法,且此方法准确、重复性好,可满足临床丙戊酸钠血药浓度测定的需求。
1材料
1.1仪器Agilent 1260型高效液相色谱仪(美国Agilent公司),包括DAD紫外检测器和自动进样器;XW-80A旋涡混合器(上海精科实业有限公司);TDZ5-WS低速多管自动平衡离心机、TG16-W高速离心机(长沙维尔康湘鹰离心机有限公司);FA-2004电子天平(常州天之平仪器设备有限公司);DCY-24S氮吹仪(青岛海科仪器有限公司);SK40-120小型超声清洗机(张家港市神科超声电子有限公司)。
1.2试剂丙戊酸钠(美国Sigma公司,批号:WXBB4060V);2-溴苯乙酮(美国aladdin公司,批号:J1222026);环己烷羧酸(美国aladdin公司,批号:J1323025);正己烷、甲醇、乙腈(天津科密欧试剂公司)均为色谱纯;三乙胺;浓氨水;浓硫酸均为分析纯;超纯水(院检验科提供);空白血清(院检验科提供)。
2方法与结果
2.1色谱条件色谱柱:ZORBAX SB-C18柱(150 mm×4.6 mm,5 μm);流动相:甲醇∶水=72∶28;检测波长:248 nm;流速:1.0 mL/min;柱温:25℃;进样量:10 μL;工作站:Agilent ChemStation。
2.2溶液配制
2.2.1丙戊酸钠储备液精密称取丙戊酸钠106.5mg至50 mL容量瓶中,加甲醇至刻度,制得2.134mg/mL的丙戊酸钠储备溶液。利用制备的丙戊酸钠储备液加入甲醇配制得1.607mg/mL、1.067mg/mL、0.803mg/mL、0.534mg/mL、0.267mg/mL、0.133mg/mL的丙戊酸钠各浓度标准品溶液。
2.2.2内标物称取环己烷羧酸50mg至100 mL容量瓶中,加0.2 mol/L的氨水稀释至刻度,再精密吸取该溶液7.5 mL至25 mL容量瓶中,加0.5 mol/L的氨水甲醇稀释至刻度,得到150 μg/mL的环己烷羧酸内标液。
2.2.3衍生化试剂精密称取2-溴苯乙酮1.0 g置于25 mL棕色容量瓶中,加乙腈定容,得40mg/mL的2-溴苯乙酮乙腈溶液。
2.2.4氨水与酸化试剂0.2 mol/L氨水配制:取浓氨水1.5 mL至100 mL的容量瓶中,加水至刻度。0.5 mol/L氨水甲醇配制:取浓氨水3.87 mL至100 mL容量瓶中,加甲醇至刻度,即得。1 mol/L硫酸配制:精密称取浓硫酸9.81 g,置于100 mL容量瓶中,加水稀释至刻度,即得。以上各试剂制备后置于4℃冰箱中保存。
2.3血样处理血液样品经3800 r/min离心5 min,取血清100 μl于离心管中,加环己烷羧酸内标液50 μL、硫酸50 μL,混匀1 min,加正己烷500 μL振荡混匀2 min,然后以5000 r/min离心5 min,取上清液有机相于干净的离心管中,下层液再加500 μL正己烷振荡混匀2 min,再次以5000 r/min离心5 min,合并两次上清有机相,加2-溴苯乙酮10 μL、三乙胺20 μL,振荡混匀2 min,于55℃水浴15 min后通氮气并继续水浴直至挥干溶剂,用甲醇100 μL溶解残渣,振荡混匀2 min,再以10 000 r/min离心5 min,取上清10 μL进样。
2.4特异性实验
2.4.1空白血清取健康人的空白血清100 μL置于离心管,然后按照步骤2.3进行处理,得色谱图A。
2.4.2空白血清+丙戊酸钠标准品+内标物取健康人的空白血清100 μL置于离心管中,加丙戊酸钠标准品10 μL,环己烷羧酸内标液50 μL,然后按照步骤2.3进行处理,得色谱图B。
2.4.3样品血清+内标物取服用丙戊酸钠患者的血清100 μL置于离心管中,然后按照步骤2.3进行处理,得色谱图C。
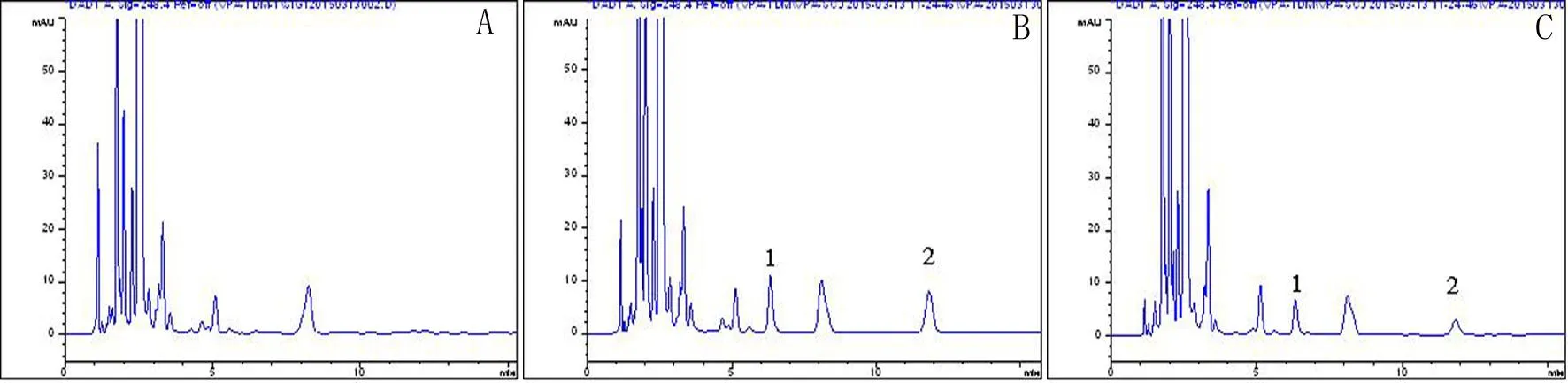
A.空白血清;B.空白血清+丙戊酸钠标准品+内标物;C.样品血清+内标物;1.环己烷羧酸(内标物);2.丙戊酸钠。
图1丙戊酸钠特异性HPLC色谱图
2.5标准曲线制备取6支2.0 mL离心管,分别加入100 μL健康人空白血清,加入2.2.1中制备的不同浓度的丙戊酸钠标准品溶液,然后按照2.3步骤进行处理,每个浓度进样2次,记录各种浓度标准品溶液中的丙戊酸钠和内标物的峰面积。以丙戊酸钠和内标物峰面积之比(R)为横坐标,以血清中丙戊酸钠终浓度(C)为纵坐标,计算丙戊酸钠线性回归方程和相关系数(r),绘制丙戊酸钠标准曲线,得到标准曲线为Y=87.79X-3.885(r=0.9979,n=6)如图2,内标物和丙戊酸钠保留时间分别约为6.3 min和11.8 min,最后结果显示丙戊酸钠血药浓度在13.3~160.7 μg/mL范围内线性关系良好。以信噪比(S/N)=6计算,丙戊酸钠最低检测限为1.0 μg/mL。
图2丙戊酸钠标准曲线图
2.6精密度试验根据2.2.1方法配制丙戊酸钠高、中、低(1.607mg/mL、0.803mg/mL、0.133mg/mL)3种浓度,然后按照2.5标准曲线的制备方法处理,得到血清中丙戊酸钠终浓度为160.7 μg/mL、80.3 μg/mL、13.3 μg/mL的样品,然后进样检测,计算并记录血清中丙戊酸钠实测浓度,每日重复测定5次,重复测定5 d,计算日内、日间RSD值,结果见表1。
表1精密度试验结果(n=5)

终浓度/(μg/mL)日内实测浓度/(μg/mL)RSD/%日间实测浓度/(μg/mL)RSD/%160.7182.8±8.44.6172.2±12.17.080.380.2±0.80.977.4±4.05.213.312.0±0.86.411.8±1.08.2
2.7回收率试验
2.7.1方法回收率取空白血清数份,分别加入浓度为1.607mg/mL、0.803mg/mL、0.133mg/mL的丙戊酸钠标准品,然后按照步骤2.3进行处理,每个浓度各做5份,根据丙戊酸钠标准曲线和线性回归方程计算各浓度标准品中丙戊酸钠的实测浓度Ct,计算丙戊酸钠实测浓度与理论浓度Cs之比,方法回收率(%)m= Ct /Cs×100%,结果见表2。
2.7.2萃取回收率取空白血清数份,分别加入浓度为1.607mg/mL、0.803mg/mL、0.133mg/mL的丙戊酸钠标准品,然后按照步骤2.3进行处理(不加内标物),每个浓度各做5份,记录各浓度标准品中丙戊酸钠峰面积R0,另取浓度为以上各丙戊酸钠标准品,加甲醇稀释制得终浓度为160.7 μg/mL、80.3 μg/mL、13.3 μg/mL的对照溶液再按照步骤2.3进行处理后,每个浓度各做5份,然后进样检测,记录各浓度对照品溶液中丙戊酸钠峰面积Rs,萃取回收率(%)e=R0/Rs×100%,结果见表2。
表2回收率试验结果(n=5)
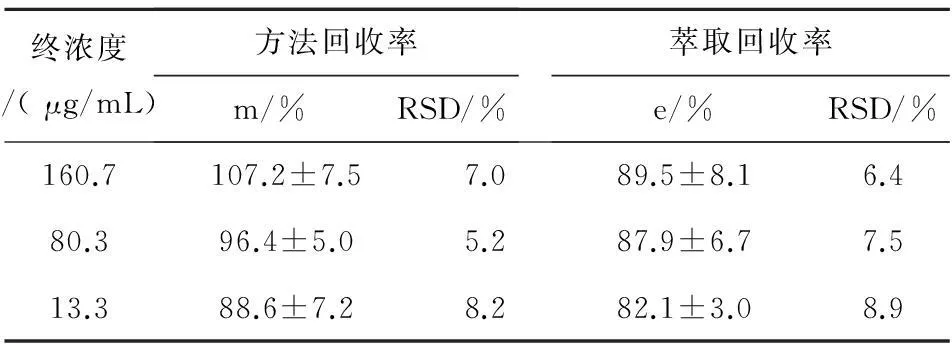
终浓度/(μg/mL)方法回收率m/%RSD/%萃取回收率e/%RSD/%160.7107.2±7.57.089.5±8.16.480.396.4±5.05.287.9±6.77.513.388.6±7.28.282.1±3.08.9
2.8专一性试验由于临床上合用药物有可能对丙戊酸钠血药浓度检测有影响,根据本实验的临床检测和药动学研究,在步骤2.1色谱条件下,对可能合用的几种药物,如苯妥英钠、卡马西平、苯巴比妥、地西泮、拉莫三嗪等进行分析,结果上述药物在本实验色谱条件下不出峰,所以以上药物对本实验测定无干扰,不会影响丙戊酸钠血药浓度的测定。
2.9稳定性试验丙戊酸钠标准品和其他相关配制溶液放置于4℃的冰箱中冷藏,分别在第1、3、5、8、11、15d时用1.067mg/mL、0.534mg/mL、0.267mg/mL丙戊酸钠标准液和其他相关溶液进行浓度测定。结果显示丙戊酸钠标准液含量未发生明显变化,说明丙戊酸储备液及相关溶液在15 d内稳定性良好。
3讨论
本实验室用甲醇-水(72∶28)为流动相,可以使主峰分离完全,峰型良好并且主峰保留时间分别为6.3 min和12.8 min较为合理,能够满足方法验证要求,同时达到节省流动相的目的。部分文献显示色谱条件为流动相甲醇-水(80∶20),检测波长235 nm、265 nm,柱温30℃时[6-7],结果不能适用本实验室临床检测。利用DAD二极管阵列检测器紫外全波段扫描显示,丙戊酸钠在衍生后在202 nm、248 nm和264 nm处有吸收,为避免与溶剂截止波长和杂质干扰,在波长248 nm处主峰吸收合理,峰型好。可知,应根据实验室具体条件:如色谱柱长短、检测器原理、流动相比例和衍生试剂的不同等因素合理选择检测波长。
由于丙戊酸钠自身在紫外条件下无吸收,必须经历酸化、萃取、衍生化后才能在紫外波长下有吸收,因此合理选择酸化剂、萃取剂和衍生化催化剂直接关系检测结果。值得一提的是本实验观察到乙醚作为萃取剂时,由于其挥发性强、毒性大等特点,与正己烷作为萃取剂相比得到的峰型较小,而正己烷可以达到萃取完全、损失少、相对安全的效果。在实验水浴催化衍生化阶段,三乙胺作为催化剂加入体积为20 μL,当增加三乙胺加入的量为50 μL和100 μL时[8-9],其实验结果并无改善,因此可选择节约的试剂成本。有文献显示实验在衍生阶段选择50℃水浴10 min[10],结果发现主峰峰面积较小,且与内标峰面积比不稳定、波动大,可能是水浴反应不完全造成。最后,本实验选择55℃水浴15 min后通入氮气并继续水浴直至溶剂挥发干,相对于自然挥发和水浴完全后再吹干,本实验采用的方法溶剂挥发完全浓缩好,且用时短一般3 min左右,降低了衍生物质接触空气被氧化变质的风险,同时节省了氮气资源。因此本法适用于本院丙戊酸钠血药浓度检测,同时实验结果可靠、准确度高、重复性好和节约实验室成本,可为临床合理用药提供参考。
【参考文献】
[1] 班立丽,唐晓霞.丙戊酸钠血药浓度与抗癫痫疗效及不良反应关系研究.中国医院用药评价与分析,2013,13(12):1086-1088.
[2] 朱斌.1214例丙戊酸钠血药浓度监测的回顾性分析.药物流行病学杂志,2008,17(1):48-49.
[3] 王宏虹,毛桂福,谭强.丙戊酸钠血药浓度监测研究概况.齐鲁药事,2012,31(10):605-607.
[4] 毛桂福,黄玉玲.HPLC测定血清中丙戊酸方法的改进.华西药学杂志,2013,28(3):305-306.
[5] 裔照国,戴月华,季中秋.对癫痫病人血清中丙戊酸钠浓度的监测.江苏药学与临床研究,2005,13(6):52-53.
[6] 展翔,徐玫.高效液相色谱法测定人血清中丙戊酸钠含量.中国药业,2009,18(6):25-26.
[7] 周莉华,涂琼,梅步云.HPLC法测定人血清中丙戊酸钠的浓度.中国药房,2011,22(26):2441-2442.
[8] 胡琳.高效液相色谱法测定丙戊酸钠的血药浓度.中国药业,2013,22(12):16-18.
[9] 李文艳,彭梅.HPLC测定人血浆中丙戊酸钠的浓度.南昌大学学报:医学版,2014,54(7):22-23.
[10] 李炳东.柱前衍生化HPLC法测定人血清中丙戊酸钠的浓度.中国美容医学,2012,21(12):195-196.
Modified high performance liquid chromatography to determine the concentration of sodium valproate in human serum
SHEN Chenjun,CHEN Xingui,WANG Yong,HU Xiuping
Department of Clinical Pharmacy,The First People′s Hospital of Chuzhou,Chuzhou 239000,China
【Abstract】Objective:To improve the high performance liquid chromatography(HPLC)for determination of the sodium valproate concentration in human serum in order to supply evidence with rational use of this drug.Methods:Cyclohexanecarboxylic acid was used as the internal standard.The serum samples were extracted with n-hexane after acidification and precipitation by sulphuric acid,and subjected to treatment with 2-bromoacetophenone as derivative reagent and triethylamine as catalytic agent.Then the samples were determined by HPLC as protocol:chromatographic column,ZORBAX SB-C18(150 mm×4.6 mm,5 μm);mobile phase,methanol-water(72∶28)at flow rate of 1.0 mL/min;detection wavelength set at 248nm;the column temperature at 25℃,and sample size in 10 μL.Results:The linear range of sodium valproate was 13.3-160.7 μg/mL(r=0.9979,n=6).The recovery rate for the sodium valproate in low,middle and high concentration and extraction recovery rate was 88.6%-107.2% and 82.1%-89.5%,respectively.The RSD of intra-day and inter-day were less than 6.4% and 8.2%,respectively.Conclusion:The modified HPLC technique can satisfy the needs of clinically monitoring the valproate concentration in patients,for it has higher specificity and accuracy as well as better linear dependence and reduced laboratory cost.
【Keywords】high performance liquid chromatography;sodium valproate;serum concentration;pharmacokinetics
文章编号:·药学·1002-0217(2016)01-0018-04
收稿日期:2015-07-24
作者简介:沈陈军(1987-),男,药师,(电话)0550-3525729,(电子信箱)jwets@126.com.
【文献标识码】【中图号】R 969.1A
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
中国现代医药杂志(2020年10期)2020-12-14
中成药(2019年12期)2020-01-04
天津医科大学学报(2019年3期)2019-08-13
中成药(2017年10期)2017-11-16
中成药(2017年10期)2017-11-16
中成药(2017年5期)2017-06-13
中国实用医药(2016年23期)2016-12-26
中国实用医药(2016年27期)2016-11-30
中国实用医药(2016年19期)2016-08-05
