
乌洛托品的热分解动力学
2016-05-09 06:35彭浩梁陈利平路贵斌张彩星周奕杉李永坚陈网桦
含能材料 2016年5期
彭浩梁, 陈利平, 路贵斌, 张彩星, 周奕杉, 李永坚, 陈网桦
(1. 南京理工大学化工学院安全工程系, 江苏 南京 210094; 2. 山西江阳化工有限公司, 山西 太原 030041)
1 引 言
乌洛托品(六亚甲基四胺)应用广泛,可用作树脂和塑料的固化剂、发泡剂、橡胶硫化的促进剂、纺织品的防缩剂等; 可作为重要的有机合成原料,用来生产氯霉素、制造农药杀虫剂等[1-2]。同时,乌洛托品可与发烟硝酸反应生成爆炸性很强的“旋风炸药”黑索今(RDX)[3],也可与醋酸、醋酸酐和硝酸铵等作为奥克托今(HMX)的合成原料[4-5]。乌洛托品属于易燃品和易制爆品。在1957年, Stranski I N等[6]就开始对乌洛托品的蒸气压和熔点进行研究; Evgeni A Gusevd等[7]采用差热分析法(DTA)对乌洛托品在密闭坩埚内的热分解行为进行了定性研究,但未对其进行深入的动力学分析与计算; Gurdip Singh等[8]利用热重测试分析了乌洛托品与高氯酸盐、锰、锌混合晶体的热分解情况,发现该混合物在高温下会发生爆炸。王鼐[9]等研究了离子液体对乌洛托品硝化制备黑索今(RDX)的影响,发现离子液体能显著影响RDX的收率,对该反应有明显的催化作用; 易文斌[10]将乌洛托品置于含氟介质中硝化制备RDX,发现在这种环境下,反应会比传统方法产生更少的废酸和硝酸盐,且此含氟相经过简单的处理就可重复使用,其催化活性不受影响。虽然前人已对乌洛托品进行了一些研究,但对其热分解行为及动力学参数计算的研究甚少。为此,本研究利用差示扫描量热仪(DSC)及绝热加速量热仪(ARC)对乌洛托品进行动态、等温和绝热测试,计算了其分解动力学参数,以期能为乌洛托品的生产、储存及运输提供参考。
2 实验样品及测试条件
乌洛托品: 纯度≥99%,白色结晶,国药集团化学试剂有限公司生产。
2.1 DSC测试
仪器: 法国赛塔拉姆(SETARAM)公司生产的差式扫描量热仪DSC131EVO。
测试条件: DSC测试所用样品池均为带镀金垫片的不锈钢高压坩埚,耐压15 MPa。动态DSC测试时,升温速率分别为1,2,4 K·min-1和10 K·min-1,温度范围为室温~350 ℃,样品质量为(1±0.02) mg; 等温DSC测试温度分别为223,226,230,233 ℃,样品质量为(3±0.02) mg。动态及等温测试的气氛均为氮气,50 mL·min-1。
2.2 2.2 ARC测试
仪器: 加速度量热仪(es-ARC),英国THT公司生产,温度测试范围: 室温~500 ℃,压力测试范围: 0~15 MPa。
测试条件:测试模式为加热-等待-搜索(HWS),加热温度梯度为5 ℃,检测灵敏度为0.02 ℃·min-1,等待时间为15 min,测试温度范围为180~340 ℃; 样品球为哈氏合金球,样品球的质量为14.008 g,比热容为0.42 J·g-1·K-1,样品质量为0.802 g。
3 实验结果与讨论
3.1 动态DSC测试
乌洛托品的动态DSC测试结果见图1。

图1乌洛托品的动态DSC曲线
Fig.1Dynamic DSC curves of urotropin
由图1可以看出,乌洛托品的动态DSC曲线中有2个放热峰、1个吸热峰,其中第一个放热峰记作W1,第二个放热峰记作W2。根据图1,1,2 K·min-1和4,10 K·min-1的吸热峰分别位于乌洛托品熔点(263 ℃)的左右两侧,所以判断吸热峰对应乌洛托品的熔融过程。由于分解和熔融过程发生重叠,因此无法获得准确的分解热和熔融热。
DSC数据的常用分析方法有Friedman法[11]、Kissinger法[12]和Ozawa法[13]等。而乌洛托品由于分解放热伴随着相变吸热,通过放热量计算热转化率的方法计算动力学参数行不通[14],故本研究采用Kissinger法计算其相关动力学参数。
Kissinger方法认为曲线峰顶温度Tmax处反应速率最大,非等温、非均相反应的动力学方程可转化为式(1):
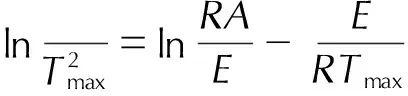
(1)
式中,β为升温速率,K·min-1,R为普适气体常数,8.314 J·mol-1·K-1,E为活化能,kJ·mol-1,A是指前因子,s-1。


图2Kissinger方法所得动力学参数的拟合结果
Fig.2Fitting results of kinetic parameters obtained from Kissinger method
3.2 等温DSC测试
3.2.1 测试结果与分析
动态DSC测试结果表明在乌洛托品的分解反应均伴随有相变,为了尽量减小相变对其分解的影响,尝试在低于动态条件下吸热信号的温度范围内(223,226,230,233 ℃)进行等温实验,测试结果见图3和表1。
表1乌洛托品等温DSC的实测结果
Table1Isothermal DSC data of urotropin
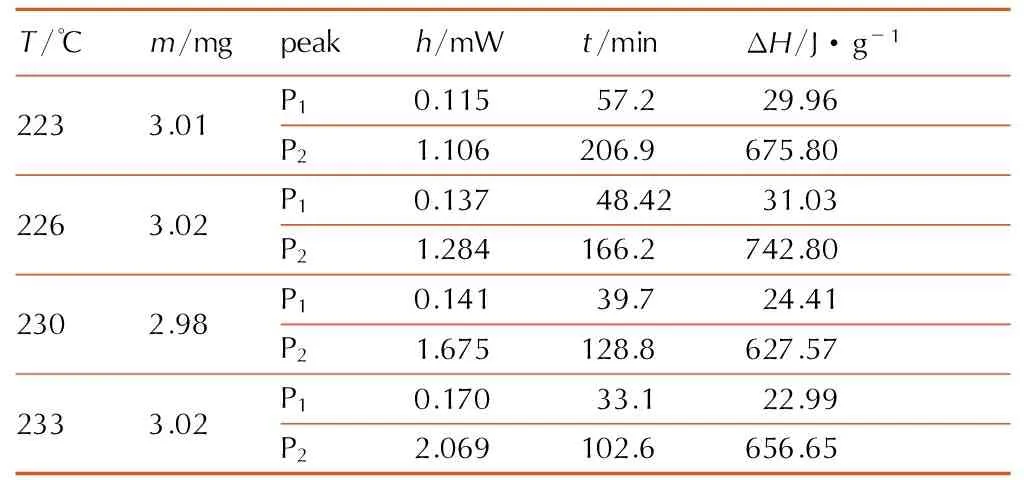
T/℃m/mgpeakh/mWt/minΔH/J·g-12233.01P10.11557.229.96P21.106206.9675.802263.02P10.13748.4231.03P21.284166.2742.802302.98P10.14139.724.41P21.675128.8627.572333.02P10.17033.122.99P22.069102.6656.65
Note: P1and P2respent the first and second exothermic peaks in isothermal test.hispeak height.tis appearance time. ΔHis decomposition heat.
由图3可以看出,乌洛托品在等温测试过程中也存在两个放热峰(P1和P2)。从图3a可以看出,其DSC曲线均为钟状,从图3b转化率曲线可以看出,该转化率曲线由两个“S”型曲线组成,据此可判断该分解反应为自催化分解反应[15]。
在等温条件下,活化能可以通过最大放热速率qm与温度T的关系求得[16-19]:
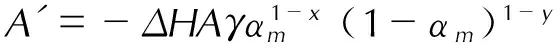
(2)
则ln(qm/D0)=lnA′-E/RT
(3)
式中,ΔH为总放热量,kJ;A为指前因子,s-1;γ为该物质最终分解的质量分数;αm表示到达最大放热速率时样品的转化率;x与y为反应机理的特征常数,在自催化反应的动力学计算中,物料的分解先加速后减速,x与加速阶段有关,y与减速阶段有关;qm为最大放热速率,mW;D0为测试样品的初始质量,mg; 基于不同温度T下的qm,以ln(qm/D0)与1/RT拟合成一条直线,根据斜率可求得E。

a. heat flow curves
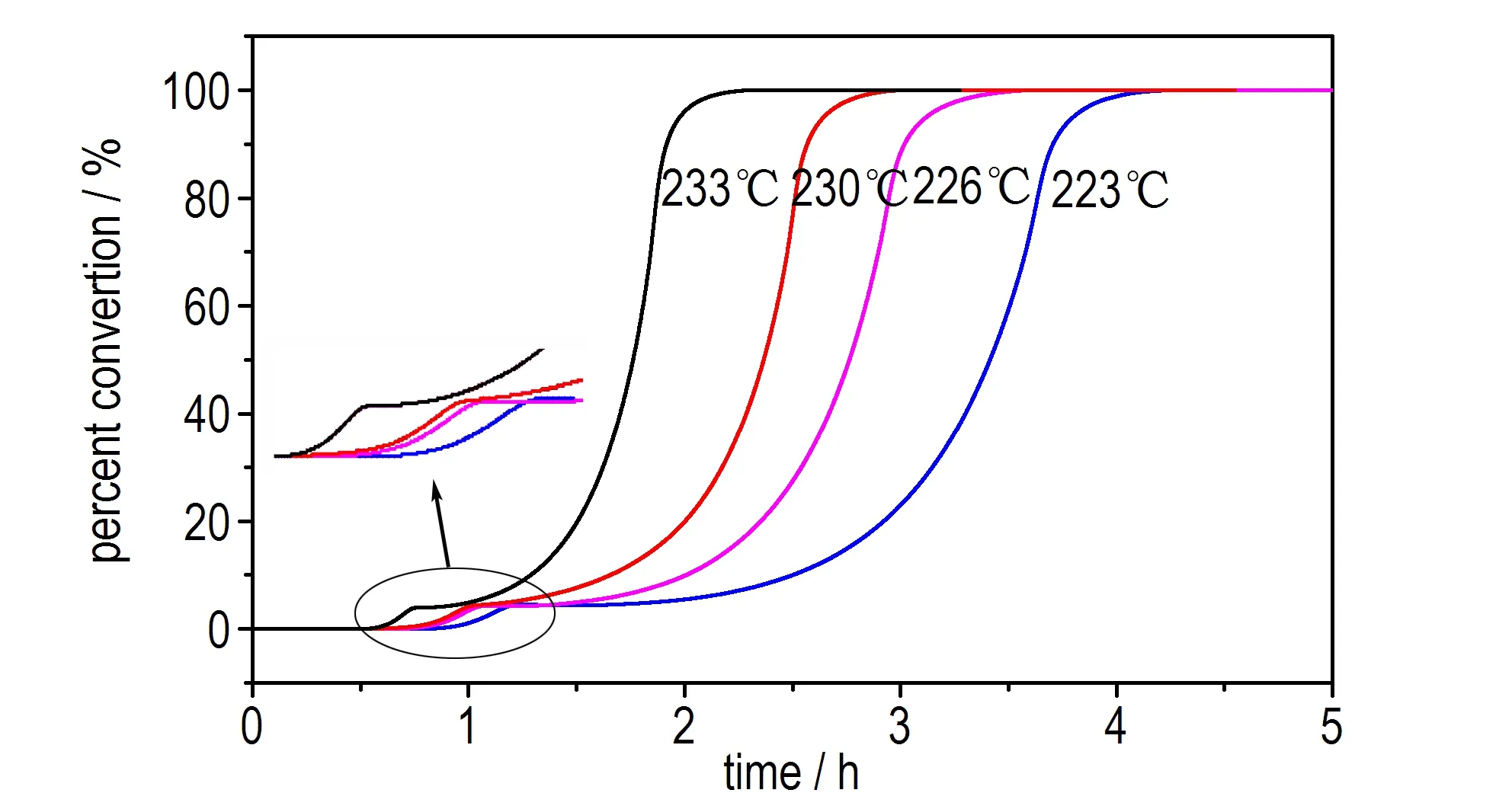
b. percent conversion curves
图3乌洛托品等温DSC测试结果
Fig.3Isothermal DSC curves of urotropin
由表1中实验所得qm和D0,结合式(3),计算得到的ln(qm/D0)与1/RT的拟合关系如图4所示。
由图4的拟合结果计算可得等温曲线中两个放热峰(P1和P2)所对应的活化能,分别为147.9 kJmol-1和153.0 kJmol-1,可见二者相差不大,所以放热峰P1、P2应是熔化吸热与分解放热相互作用的结果。
为了检验该结果的准确性,采用Friedman法进行了活化能的计算,得到等温过程中两个峰对应的E随转化率的变化情况,如图5所示。
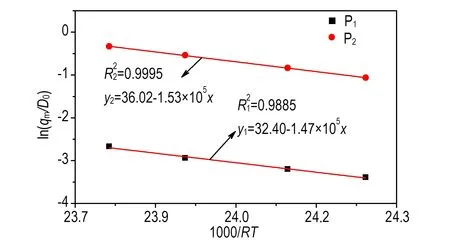
图4乌洛托品的ln(qm/D0)与1/RT关系曲线
Fig.4Curves of ln(qm/D0) vs 1/RTfor urotropin
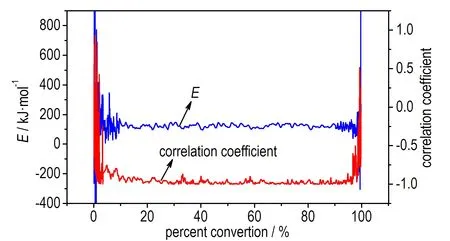
a. E and correlation coefficient vs α of P1
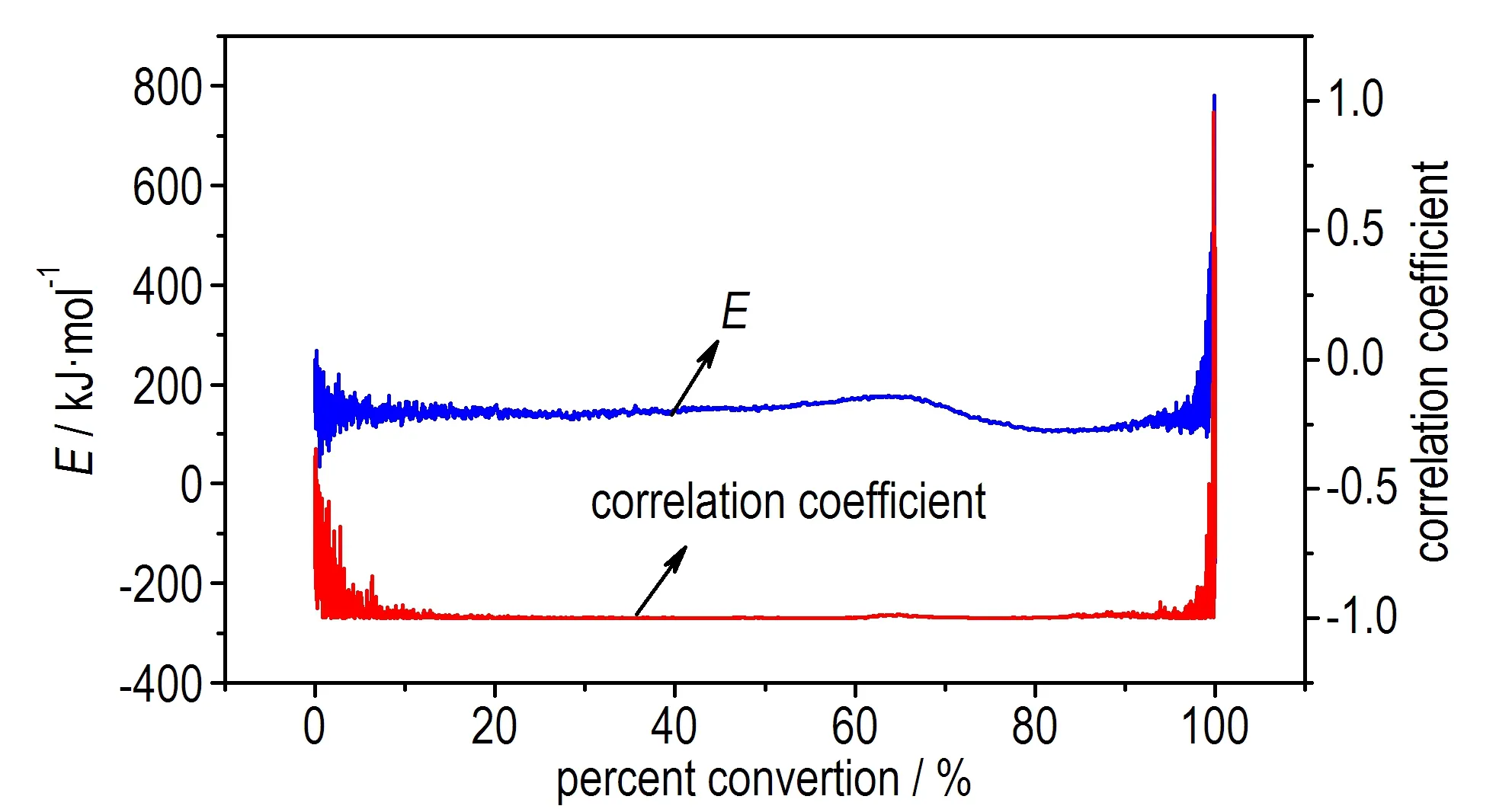
b. E and correlation coefficient vs α of P2
图5P1和P2的活化能和线性回归相关系数随α的变化曲线
Fig.5Eand correlation coefficient vsαof P1and P2
由于在α<0.1和α>0.9的范围内,即在反应的开始及结束阶段,基线影响较大,且容易受到仪器噪音的影响,因此取0.1<α<0.9作为分析对象。结果发现,由Friedman法得到放热峰P1和P2的活化能E基本都保持在150 kJ·mol-1左右,且线性回归相关性高。根据文献[16]的判别方法,进一步表明P1和P2的反应很可能遵循同一反应机理,是分解放热与相变吸热相互竞争导致,这与利用等温测试中的最大放热速率计算得到的结果一致。
3.2.2 等温DSC数据推导TD24
在上述分析的基础上采用瑞士AKTS公司开发的专门研究物质分解热动力学及热安全性的分析软件(AKTS),对乌洛托品的等温DSC数据进行动力学分析,在此基础上预测绝热条件下最大温升速率到达24h时所对应的温度TD24,以便为生产、储存和运输工艺温度的确定提供参考。结果见图6,从图6中可知TD24为216.26 ℃。

图6AKTS软件预测的乌洛托品绝热诱导期(TMR)曲线
Fig.6TMRcurves predicted by AKTS for urotropin
3.3 ARC测试
3.3.1 测试结果与分析
为了更全面地评估乌洛托品的热分解特性,同时验证AKTS软件对TD24参数预测结果的准确性,采用ARC测试乌洛托品在绝热条件下的热分解,并求解其TD24。ARC实验中获得样品的时间-温度曲线、时间-温升速率曲线、温度-压力曲线及温度-温升速率曲线如图7,获得的热分解参数见表2。
从图7a和7c中可以看出,样品在230 ℃左右分解,在分解过程中压力随着温度的升高而增大,且约0.5 h后温度和压力急剧上升,尤其在470~480 min的10 min内由235 ℃(1.5 MPa)骤增至270 ℃(2.5 MPa),显示出了热爆炸特性,危险性较大。图7b和图7d曲线显示在分解开始后不久温升速率出现了较大的波动(图中圈出位置),推测可能是因为在该时间段乌洛托品发生了熔化,导致热量波动,这与DSC的测试结果一致。
文献[20-21]报道,绝热体系中Townsend提出的绝热动力学计算模型最常用,该模型基于n级分解动力学,认为绝热体系中温升速率与温度的关系为:
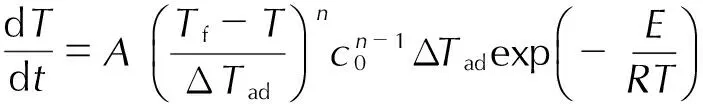
(4)

(5)
将其代入式(4)并取对数得:
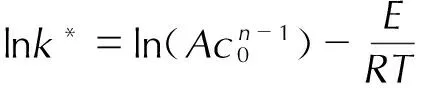
(6)
式中,T为温度,K;t为时间,min;Tf为绝热条件下达到的最高温度,K; ΔTad为绝热温升,K;n为反应级数;c0为反应物起始浓度,mol·L-1。
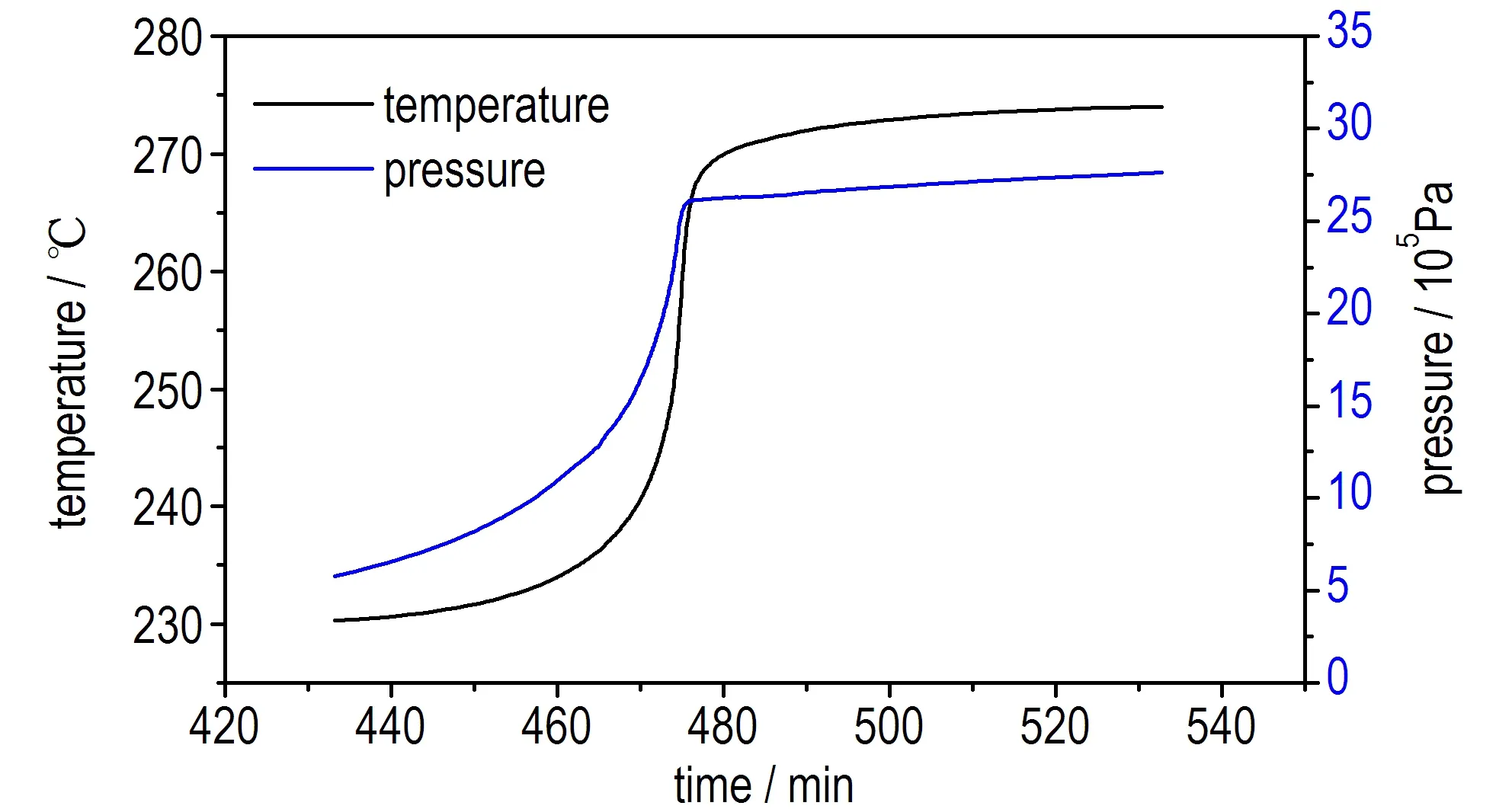
a. temperature vs time

b. dT/dt vs time

c. pressure vs temperature
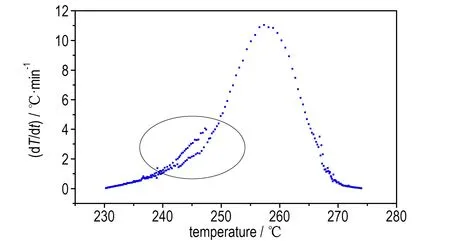
d. dT/dt vs temperature
图7乌洛托品ARC测试结果
Fig.7Test results of ARC for urotropin
表2乌洛托品热分解特征参量
Table2Measured thermal decomposition characteristic parameters for urotropin

initialtemperature/℃initialtemperaturerate/℃·min-1maxratetemperature/℃maxtemperaturerate/℃·min-1maxpressurerate/MPa·min-1finaltemperature/℃maxpressure/MPaadiabatictemperaturerise/℃230.280.023257.3511.040.353273.892.74743.61
利用式(4),将n、E、A作为未知数进行非线性拟合得到该分解反应的反应级数为3,活化能为473.6 kJ·mol-1,指前因子为1.47×1045。将此反应级数带入式(6)进行验证,发现线性拟合效果较好,相关系数达到0.991,具体的拟合结果见图8。
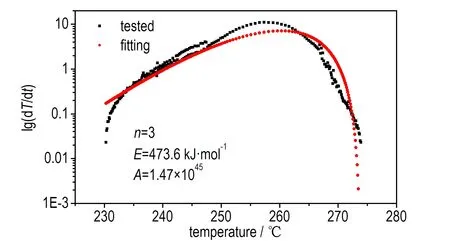
a. nonlinear fitting by Townsend method

b. confirmation of fitting result
图8使用Townsend方法求取动力学参数的拟合曲线
Fig.8Fitting curves of kinetic parameters obtained by Townsend method
从图8b可以看出,反应的开始、结束阶段的线性度较差,可能是测试开始时存在一定的误差,出现热量波动; 也可能是样品的分解较复杂,未能遵循一个反应机理。这里选取中间线性度比较好的部分进行拟合,拟合结果线性相关系数较高,因此,可以认为计算所得的活化能较可靠,可以用此数值进行下一步最大反应速率到达时间的计算。
从测试结果可以看出,由Townsend方法计算所得的乌洛托品样品的表观活化能(473.6 kJ·mol-1)较大。原因是Townsend方法主要依赖于温升速率来计算活化能,而ARC测试时的样品球质量(14.008 g)远大于样品质量(0.802 g),测试反应中释放的热量有一部分不可避免地用来加热样品球,使得实测的温升速率是经过样品球“钝化”后的结果,即综合了样品分解放热、样品球吸热以及传热等因素,不能代表样品的真实温升速率。因此,分析样品在绝热条件下温度与最大反应速率到达时间的关系时,必须加以修正。
3.3.2 测试结果的修正
为了减小用于加热样品球的那部分热量对反应的影响,此处引入热修正系数Ф进行修正:

(7)
式中,Ms为试样质量,g;cvs为试样平均比热容,J·g-1·k-1;Mb为试样容器质量,g;cvb为试样容器的比热容,J·g-1·k-1。计算中所用乌洛托品比热容为1.24 J·g-1·k-1,由THT公司生产的微反应量热仪(uRC)测得,计算得Φ值为6.93。根据文献[21],最大反应速率到达时间满足以下关系:

(8)
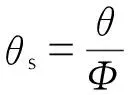
(9)
式中,θ为实测最大反应速率到达时间,s;θs为样品真实的最大反应速率到达时间,s。由式(8)和(9)计算得TD24为212.5 ℃,这与基于等温DSC数据的预测结果(216.26 ℃)基本一致,因此认为该值具有一定的参考价值。
4 结 论
(1) 乌洛托品的动态DSC测试结果表明,分解放热和熔化吸热重叠,通过Kissinger法分别对两个放热峰进行拟合计算,得到两个峰的表观活化能分别为90.62 kJ·mol-1和99.77 kJ·mol-1。
(2) 乌洛托品的等温DSC曲线同样显示两个放热峰,且这两个放热峰的表观活化能基本一致,表明这两个放热峰的反应机理相同,即两个放热峰属于同一个反应,只是由于叠加了熔融吸热峰,导致两个放热峰的形成。同时,利用AKTS软件基于等温DSC数据预测得到乌洛托品绝热条件下的TD24为216.26 ℃。
(3) 绝热量热测试得到乌洛托品的起始分解温度为230.28 ℃,且放热反应剧烈,产气量较大,属于分解发生便很难控制的一类物质。认为绝热测试中温升速率的波动也是由于熔融吸热所致。由绝热数据计算得到样品的TD24为212.5 ℃,与基于等温DSC数据的预测结果基本一致。
参考文献:
[1] 刘乾开, 朱国念. 新编农药使用手册(第二版)[M]. 上海: 上海科学技术出版社, 1999: 309.
LIU Qian-kai, ZHU Guo-nian. Pesticide use manual(second edition)[M]. Shanghai Scientific and Technical Publishers, 1999: 309.
[2] 李薇, 陈天科, 徐辉, 等. 乌洛托品的气相色谱分析[J]. 分析仪器, 2007(3): 29-31.
LI Wei, CHEN Tian-ke, XU Hui, et al. Gas chromatographic analysis of urotropin[J].AnalyticalInstrumentation, 2007(3):29-31.
[3] WANG S, PANG R, ZHANG Z. Determination of formaldehyde in pharmaceutical product ofurotropine[J].ChineseJournalofPharmaceuticalAnalysis, 2010, 30(9): 1774-1776.
[4] 李全良. 奥克托今的合成工艺研究[D]. 太原:中北大学, 2007.
LI Quan-liang. Study on the technology of synthesis of HMX[D]. Taiyuan: North university of China, 2007.
[5] 李伟明. 醋酐法生产HMX工艺研究[D]. 太原: 中北大学, 2009.
LI Wei-ming. Study on process of HMX by acetic anhydride method[D]. Taiyuan: North university of China, 2009.
[6] Stranski I N, Klipping G, Bogenschuetz A F, et al. Thermal decomposition of hexamethylenetetramine[J].AdvancesinCatalysis, 1957(9): 406-414.
[7] Gusev E A, Dalidovich S V, Krasovskaya L I. Investigation of urotropine thermal decomposition reaction in self-generated atmosphere by means of thermal analysis method[J].ThermochimicaActa, 1985, 93: 21-24.
[8] Singh G,Baranwal B P, Kapoor I P S, et al. Preparation, X-ray crystallography, and thermal decomposition of some transition metal perchlorate complexes of hexamethylenetetramine[J].TheJournalofPhysicalChemistryA, 2007, 111(50): 12972-12976.
[9] 王鼐, 石煜, 杨红伟, 等. 离子液体对乌洛托品硝解反应的影响[J]. 含能材料, 2011, 19(3): 252-257.
WANG Nai, SHI Yu, YANG Hong-wei, et al. Nitrolysis of hexamethylenetetramine in presence of ionic liquids[J].ChineseJournalofEnergeticmaterials, 2011, 19(3): 252-257.
[10] 易文斌, 蔡春. 含氟介质中乌洛托品硝解制 RDX 研究[J]. 含能材料, 2008, 16(6): 679-681.
YI Wen-bin, CAI Chun. Preparation of RDX by nitrolysis of hexamethylenetetramine in fluorous media[J].ChineseJournalofEnergeticMaterials, 2008, 16(6): 679-681.
[11] Friedman H L. Kinetics of thermal degradation of char-forming plastics fromthermogravimetry. Application to a phenolic plastic[J].JournalofPolymerSciencePartC:PolymerSymposia, 1964, 6(1): 183-195.
[12] Kissinger H E. Reaction kinetics in differential thermal analysis[J].AnalyticalChemistry, 1957, 29(11): 1702-1706
[13] Hansen L D,Lewi E A, Eatough D J, Bergstrom R G, et al. Kinetics of drug decomposition by heat conduction calorimetry[J].PharmaceutRes, 1989, 6(1): 20-27.
[14] Zhang C X, Lu G B, Chen L P, et al. Two decoupling methods for non-isothermal DSC results of AIBN decomposition[J].JournalofHazardousMaterials, 2015, 285: 61-68.
[15] Ozawa T. A new method of analyzingthermogravimetric data[J].BulletinoftheChemicalSocietyofJapan, 1965, 38(11): 1881-1886.
[16] Li X R,Koseki H. Thermal decomposition kinetic of reactive solids based on isothermal calorimetry measurements[J].JThermAnalCalorim, 2006, 85(3): 637-642.
[17] Hansen L D,Lewi E A, Eatough D J,et al. Kinetics of drug decomposition by heat conduction calorimetry[J].PharmaceutRes, 1989, 6(1): 20-27.
[18] Li X R,Koseki H. SADT prediction of autocatalytic material using isothermal calorimetry analysis[J].ThermochimActa, 2005, 431(1): 113-116.
[19] 彭敏君. 两种偶氮化合物热危险性及热分解机理研究[D]. 南京:南京理工大学, 2014.
PENG Min-jun. Study on thermal hazards and decomposition mechanisms of two azo compounds[D]. Nanjing :Nanjing University of Science And Technology, 2014.
[20] 陈利平. 甲苯硝化反应热危险性的实验与理论研究[D]. 南京: 南京理工大学, 2009.
CHEN Li-ping. Experimental and theoretical studies on thermal hazards of toluenenitrations[D]. Nanjing:Nanjing University of Science And Technology, 2009.
[21] Townsend D I, Tou J C. Thermal hazard evaluation by an accelerating rate calorimeter[J].ThermochimicaActa, 1980, 37(1): 1-30.
猜你喜欢
空气动力学学报(2022年4期)2022-08-23
黑龙江大学自然科学学报(2022年1期)2022-03-29
中学生数理化·高一版(2020年6期)2020-07-25
中学生数理化·八年级物理人教版(2017年3期)2017-11-09
信息安全与通信保密(2016年3期)2016-08-23
中国人兽共患病学报(2016年6期)2016-01-30
中国塑料(2015年3期)2015-11-27
浙江大学学报(工学版)(2015年2期)2015-05-30
制造技术与机床(2015年3期)2015-01-27
中国塑料(2014年2期)2014-10-17
