
1,2,4,5-四嗪的四唑类衍生物高能量密度材料的分子设计
2016-05-09 06:37:21宋纪蓉马海霞
含能材料 2016年5期
陈 沫, 宋纪蓉, 马海霞
(1. 西北大学化工学院, 陕西 西安 710069; 2. 北京故宫博物院文保科技部, 北京 100080)
1 引 言
四嗪类高氮化合物是近年发展起来的一类具有良好应用前景的新型含能材料,其分子结构中含有较多的N—N和C—N键,四嗪环含氮量高达68.3%,具有高能量、热稳定性好、低特征信号、燃烧无残渣及无污染等优点,广泛应用于推进剂、新型高能钝感炸药和烟火剂等含能材料领域[1-14]。目前的研究主要是发现和合成可以取代环三亚甲基三硝胺(RDX)和1,3,5,7-四硝基-1,3,5,7-四氮杂环辛烷(HMX)的四嗪化合物,实验中研究较多的四嗪类化合物有: 3,6-二氨基-1,2,4,5-四嗪-1,4-二氧化物(LAX-112)[15]、3,3′-偶氮-(6-氨基-1,2,4,5-四嗪)(DAAT)[16]、3,6-二肼基-1,2,4,5-四嗪(DHTz)[17]、3,6-双(1-氢-1,2,3,4-四唑-5-氨基)-1,2,4,5-四嗪(BTATz)[18]、3,6-双硝基胍基-1,2,4,5-四嗪(DNGTz)[19]、3,6-二(3,5-二甲基吡唑-1-基)-1,2,4,5-四嗪(BT)[20]等。
四唑环是一种五元氮杂环,是唑类化合物中研究较多的一类,由于H原子位置的变动,可以分为三种同分异构体,即1H-四唑、2H-四唑和5H-四唑,而1H-四唑是目前研究较多的一类。四唑环含氮量高(80%),具有高正生成焓,可以用于研究光敏材料及燃料电池,也可以制备起爆药和固体推进剂等含能化合物,还可以作为多种反应中的催化剂,例如Suzuki、Heck等偶联反应,应用较为广泛[21-25]。四唑环上的所有原子共平面,具有芳香性,稳定性好,因此是开发新型含能化合物的理想基团[26,27]。四唑环可以通过各种反应取代环上的H原子来接多种含能基团以改善物质的性能,如硝基(—NO2)、偶氮基(—NN—)和氰基(—CN)等基团,可以很好地增加分子内氮含量及能量密度。
本研究选择能量较高的1,2,4,5-四嗪环为基本骨架,结合四唑环设计了30种1,2,4,5-四嗪衍生物,筛选出wB97/6-31+G**作为研究该类化合物的方法和基组,在该水平下对这些化合物进行理论计算研究,采用原子化方案获得其标准摩尔生成焓,并在生成焓和密度基础上预测爆轰性能,以对新型四嗪含能物的设计和性能研究提供参考。
2 计算方法
运用DFT方法,在B3LYP/(6-31G*,6-311G*,6-31+G*,6-31G**,6-311G**,6-31+G**,6-311++G**,cc-pVDZ,cc-pVTZ)水平下计算分析1,2,4,5-四嗪、3,6-二氨基-1,2,4,5-四嗪、3,6-二肼基-1,2,4,5-四嗪和3,6-二叠氮基-1,2,4,5-四嗪4种化合物生成焓,通过与实验值进行对比拟合,在B3LYP/6-31+G**水平下计算的结果与实验值线性相关性最好,达到0.9863,所以选择基组6-31+G**和不同方法(B3PW91,M05,M05-2X,M06,M06-2X,wB97)组合计算上述4种化合物生成焓,通过与实验值进行对比拟合,在wB97/6-31+G**水平下计算的结果与实验值线性相关性最好,达到0.9896。在wB97/6-31+G**水平下对所设计的30种1,2,4,5-四嗪衍生物的几何结构进行全优化,经振动频率分析表明优化构型为势能面上极小点(无虚频),得到的热力学数据采用原子化方案(atomization scheme)[28-32]预测目标化合物的标准生成焓。首先将分子分解为原子:
CaHbOcNd(g)→aC(g)+bH(g) +cO(g) +dN(g)
(1)
则该反应在298 K时的标准反应焓ΔH298由(2)式计算:
ΔH298=ΣΔHf,p-ΣΔHf,R=
aΔHf,C+bΔHf,H+cΔHf,O+dΔHf,N-ΔHf,CaHbOcNd
(2)
式中,ΔHf,R和ΔHf,p分别表示反应物和生成物在298 K的标准生成焓; ΔHf,C、ΔHf,H、ΔHf,O和ΔHf,N分别为原子C、H、O和N在298 K的标准生成焓,可从手册中查得; ΔHf,CaHbOcNd为分子CaHbOcNd在298 K的标准生成焓,为待求项。同时存在下列关系式:
ΔH298=ΔE298+Δ(pV) =ΔE0+ΔEZPE+ΔET+ΔnRT
=E0,C+E0,H+E0,O+E0,N-E0,CaHbOcNd-
EZPE,CaHbOcNd-ΔET,CaHbOcNd+ΔnRT
(3)
式中,E0,C、E0,H、E0,O、E0,N和E0,CaHbOcNd分别为wB97/6-31+G**水平下计算得到的原子C、H、O、N和分子CaHbOcNd在0 K的总能量;EZPE,CaHbOcNd和ΔET,CaHbOcNd分别为分子CaHbOcNd的零点能和热校正值,可从振动分析获得的热力学数据得到,对原子而言,EZPE和ΔET项均为0; Δn表示气体产物和反应物的物质的量之差,R是气体常数,8.314 J·mol-1·K-1;T表示绝对温度,K。综合上式,化合物CaHbOcNd在298 K的标准生成焓ΔHf,CaHbOcNd即可求得。
运用Kamlet和Jacobs于1968年提出的半经验K-J方程[30-36]估算其爆速(D)、爆压(p)值:
D=Φ0.5(1.011+1.312ρ)
(4)
(5)
p=1.558Φρ2
(6)

化学键强度可用键离解能(EBD)来衡量,它对理解化学反应过程十分重要[37]。在UwB97/6-31+G**水平下计算标题化合物的EBD,对比化合物键的强度及热稳定性。在298 K和1个标准大气压压下,化学键A—B均裂所需能量等于反应A—B(g)→A·(g)+B·(g)的反应焓,该反应焓可定义为A—B的键离解焓[38]。文献报道许多有机化合物的键离解能和键离解焓是相等的[39-42]。因此,标题化合物的键离解能可依据方程式(7)进行计算[39]:
EBD(A-B)=H(A·)+H(B·)-H(A-B)
(7)
本研究所有化合物密度均采用摩尔体积法(ρ=M/Vm)计算得到,其中M为化合物的摩尔质量,Vm为化合物的摩尔体积,是在稳定构型下,基于0.001e·bohr-3等电子密度面所包围的体积空间,用Monte-Carlo方法求得。所有计算使用Gaussian09[43]量子化学软件包完成。
3 结果讨论
3.1 几何结构
选择能量较高的1,2,4,5-四嗪环作为母体,结合四唑环、偶氮基(—NN—)以及硝基(—NO2)设计了30种四唑类1,2,4,5-四嗪衍生物,分子结构如图1所示。在wB97/6-31+G**水平下对30种1,2,4,5-四嗪衍生物进行几何全优化,表1列出了标题化合物中部分对称双取代化合物部分几何参数(键长、键角和二面角),计算结果表明,1,2,4,5-四嗪中H原子被四唑环取代后,四嗪环上各键(C—N、N—N)键长均受到影响,化合物iii1~iv3中与四唑环相邻的四嗪环上C—N键长依次为0.1333/0.1334,0.1329/0.1330,0.1333,0.1333/0.1336,0.1332/0.1335和0.1333/0.1335 nm,略小于未被取代的1,2,4,5-四嗪中C—N键长0.1336 nm(实验值0.1338 nm),减幅为0~0.0007 nm之间; 连接两环间偶氮基(—NN—)的引入使四嗪环上与取代基相邻的C—N键长略增加; 四唑环上引入硝基—NO2和偶氮硝基—NN—NO2使环上C—N键长略减小,N—N键长略增加。四嗪环上所有C—N键的键长在0.1329~0.1336 nm之间,N—N键长约为0.1303~0.1310 nm,键长趋于均匀化,C—N键键长短于标准C—N单键(0.1470 nm)而长于标准CN双键(0.1280 nm)的键长; 同样,N—N键也短于标准N—N单键(0.1450 nm)长于标准NN双键(0.1250 nm),四嗪环中六个p电子形成大π键,而非三个首尾相接的双键,表明1,2,4,5-四嗪衍生物的四嗪环具有一定的芳香性。
与未取代的1,2,4,5-四嗪比较,化合物iii1~iv3的四嗪环内键角N(2)C(3)N(4)和N(5)C(6)N(1)增大,其余键角减小,且环内键长和键角差异都较小,这是因为化合物具有较好的对称性和共轭性。化合物iii1,iii2和iii3的二面角N(1)C(6)N(7)N(9)和N(5)C(6)N(7)C(12)分别为172.96°和173.57°,115.85°和122.71°, 8.66°和24.97°,iii1的接近180°,所以iii1的四嗪环和四唑环具有较好的共面性。化合物iv1,iv2和iv3的二面角N(5)C(6)N(7)N(9)和N(2)C(3)N(8)N(10)分别为-141.63°和-140.63°,-40.92°和-139.79°,40.64°和142.29°,二面角C(6)N(7)N(9)N(11)和C(3)N(8)N(10)N(12)分别为-178.49°和2.00°,164.82°和14.39°,0.46°和11.32°,表明四嗪环与四唑环不共面,连接桥—NN—的引入使四嗪环与四唑环共面性更差。

图11,2,4,5-四嗪衍生物的分子结构
Fig.1Molecular structures of 1,2,4,5-tetrazine derivatives
表1部分标题化合物的键长、键角和二面角
Table1Calculated bond lengths, bond angles and dihedral angles of part of the title compounds

parametersatomiii1iii2iii3iv1iv2iv3s-tetrazinebondlength/nmR(1,2)0.13060.13080.13090.13070.13070.13070.1313R(2,3)0.13330.13290.13290.13350.13350.13350.1336R(3,4)0.13340.13300.13300.13350.13330.13330.1336R(4,5)0.13060.13080.13030.13090.13100.13100.1313R(5,6)0.13330.13290.13330.13330.13320.13330.1336R(6,1)0.13340.13300.13330.13360.13350.13350.1336R(6,7)0.14030.14190.14000.14230.14240.1425R(7,9)0.13560.13470.13640.12380.12350.1234R(7,12)0.13590.13490.1359R(9,10)0.12750.12840.1269R(10,11)0.13640.13530.1368R(11,12)0.13040.12980.1299R(9,11)0.13660.13770.1368R(11,13)0.13490.13470.1354R(11,16)0.13620.13580.1359R(13,14)0.12730.12750.1274R(14,15)0.13720.13680.1368R(15,16)0.13020.12930.1299bondangle/(°)A(1,2,3)116.13115.97116.67116.71116.55116.69116.98A(2,3,4)126.37127.54126.90126.34126.73126.60126.04A(3,4,5)117.48116.49116.31116.78116.59116.58116.98A(4,5,6)116.13115.97116.69116.69116.53116.70116.98A(5,6,1)126.37127.54126.72126.41126.77126.59126.04A(6,1,2)117.48116.49116.07116.78116.61116.62116.98
(Table1Continued)

parametersatomiii1iii2iii3iv1iv2iv3s-tetrazinebondangle/(°)A(8,3,2)118.10116.37117.48114.54114.37114.32A(8,3,4)115.53116.08115.62118.95118.75118.92A(5,6,7)118.10116.37114.96114.44118.93118.93A(1,6,7)115.53116.08118.32119.01114.17114.35A(6,7,9)122.85121.14123.20111.59111.90110.82A(6,7,12)129.09131.91128.40A(7,9,10)106.09106.67106.52A(9,10,11)111.73111.56112.06A(10,11,12)106.04105.08105.13A(11,12,7)108.09109.93109.51A(12,7,9)108.05106.76106.73A(9,11,16)132.92134.26123.74A(9,11,13)118.32118.42128.32A(11,13,14)106.00106.59105.72A(13,14,15)111.51111.61112.35A(14,15,16)106.29105.20105.12A(15,16,11)107.43109.36108.89A(16,11,13)108.76107.24107.88dihedralangle/(°)D(1,6,7,9)172.96115.858.6642.40142.92-143.17D(5,6,7,12)173.57122.7124.97D(2,3,8,16)173.57-122.71132.20D(4,3,8,13)172.96-115.85125.00D(1,6,7,12)-5.95-58.46-154.61D(5,6,7,9)-7.52-62.99-171.76-141.63-40.9240.64D(2,3,8,10)-140.63-139.79142.29D(6,7,9,11)-178.45177.30-178.64D(3,8,10,12)-177.93-177.37178.82D(7,9,11,13)-178.49164.82 0.46D(8,10,12,17) 2.0014.3911.32
Note: These data in the brackets are atom number.
3.2 生成焓
生成焓是化合物的基本热力学性质,是其能量高低的标志,也是衡量高能量密度材料(HEDM)爆轰性能的重要参数,因此精确计算生成焓对设计、筛选以及合成新型品优HEDM具有重要意义。本研究在wB97/6-31+G**水平下采用原子化方案估算1,2,4,5-四嗪衍生物及传统含能材料RDX和HMX的生成焓,表2列出了各化合物的总能量(E0)、零点能(EZPE)、温度校正值(ET)、含氮量(N%)及生成焓(ΔHf)。计算结果表明,所有1,2,4,5-四嗪衍生物生成焓均大于传统含能材料RDX和HMX,具有高正生成焓,其中最高的生成焓值为2610.45 kJ·mol-1。四唑环有利于提高四嗪衍生物的ΔHf,如化合物i1和iii1,且四唑环上引入—NO2和—NN—NO2能够进一步改善四嗪衍生物的生成焓,比其它衍生物(如i1和iii1)生成焓高,且基团数量越多,生成焓越高,说明四唑环上引入—NO2和—NN—NO2能够有效地改善四嗪衍生物的ΔHf。含氮量越高,生成焓不一定越大,如化合物iv1的含氮量(81%)最高,生成焓为1920.40 kJ·mol-1。
图2对比不同取代基对四嗪衍生物生成焓的影响,结果表明对称双取代化合物的生成焓明显高于相应单取代的,且四唑环上引入—NN—NO2能够显著提高四嗪衍生物的生成焓。不同系列衍生物与氮原子数的关系如图3所示,从图3可以看出,同系列化合物的生成焓随氮原子数量的增加而逐渐增大,且相同氮原子数量的四嗪衍生物生成焓不同但相近。综上所述,四唑环及在其上引入—NO2和—NN—NO2在增加1,2,4,5-四嗪衍生物的生成焓方面起了非常重要的作用,且同系列1,2,4,5-四嗪衍生物的生成焓随氮原子数的增加而逐渐增大,含氮量越高,生成焓不一定越大。
3.3 前线轨道能量
基于分子轨道理论,化合物的最高占据轨道能量(EHOMO)越低,最低空轨道能量(ELUMO)越高,即分子轨道能级差ΔE=ELUMO-EHOMO越大,化合物越稳定。
表21,2,4,5-四嗪衍生物及RDX和HMX的总能量(E0)、零点能(EZPE)、温度校正值(ET)和生成焓(ΔHf)计算值
Table2Calculated total energies (E0), zero-point energies (EZPE), thermal corrections (ET) and enthalpies of formation(ΔHf)of 1,2,4,5-tetrazine derivatives together with RDX and HMX

compd.formulaE0/HartreeEZPE/HartreeET/HartreeN/%ΔHf/kJ·mol-1i1C3H2N8 -553.31980.08030.008574.7 920.46i2C3N10O4 -962.23970.08550.013658.3 1045.69i3C3N14O4 -1181.09830.10390.017766.2 1580.41i4C3N12O4 -1071.66620.09460.015662.7 1319.86i5C3N12O4 -1071.67220.09480.015762.7 1305.09i6C3HN9O2 -757.77790.08320.011064.6 988.48i7C3HN9O2 -757.78350.08260.011164.6 972.74i8C3HN11O2 -867.20390.09240.012969.1 1264.36i9C3HN11O2 -867.21540.09210.013269.1 1233.96ii1C3H2N10 -662.73770.09020.010278.7 1219.16ii2C3N12O4 -1071.65490.09490.015662.7 1350.39ii3C3N16O4 -1290.51180.11300.019669.1 1888.81ii4C3N14O4 -1181.08440.10410.017566.2 1616.96ii5C3N14O4 -1181.08700.10420.017766.2 1610.79ii6C3HN11O2 -867.19200.09270.012969.1 1296.36ii7C3HN11O2 -867.20160.09250.012969.1 1270.36ii8C3HN13O2 -976.62650.10200.014972.5 1550.48ii9C3HN13O2 -976.63350.10170.015072.5 1531.42iii1C4H2N12 -810.35050.10800.012077.1 1322.77iii2C4N14O4 -1219.26400.11330.017163.6 1465.14iii3C4N18O4 -1438.12230.13170.021069.2 2000.01iii4C4N16O4 -1328.69070.12250.019166.7 1738.84iii5C4HN13O2 -1014.80820.11070.014669.2 1391.53iii6C4HN15O2 -1124.23340.11980.016572.2 1669.24iv1C4H2N16 -1029.18570.12730.015681.8 1920.40iv2C4N18O4 -1438.09390.13220.021169.2 2075.90iv3C4N22O4 -1656.95230.15020.025173.3 2610.45iv4C4N20O4 -1547.51930.14120.022971.4 2352.78iv5C4HN17O2 -1233.64110.12980.018374.7 1995.03iv6C4HN19O2 -1343.06580.13870.020376.7 2273.37RDXC3H6N6O6-897.35320.14700.013437.8244.21HMXC4H8N8O8-1196.47500.19720.017837.8317.19

图2均四嗪衍生物生成焓的比较
Fig.2Compared the ΔHfof s-tetrazine derivatives
本研究在几何优化基础上,获得1,2,4,5-四嗪衍生物EHOMO及ELUMO,进一步计算得到分子轨道能级差ΔE,列于表3。通过比较1,2,4,5-四嗪衍生物i1(0.33645a.u.)和i2(0.34241a.u.)、iii1(0.33575a.u.)和iii2(0.34138a.u.)能级差可以看出,当四唑环上H被—NO2取代后,EHOMO和ELUMO均减小,能级差增大,说明—NO2有利于提高1,2,4,5-四嗪衍生物的稳定性。通过i1(0.33645a.u.)和ii1(0.33331a.u.)、iii1(0.33575a.u.)和iv1(0.32594a.u.)能级差可以看出,连接桥偶氮基—NN—使1,2,4,5-四嗪衍生物EHOMO和ELUMO增大,能级差减小,且iv3偶氮基最多,能级差最小(0.31803a.u.),说明—NN—不利于1,2,4,5-四嗪衍生物稳定性的提高。i2(0.34241a.u.)和iii2(0.34138a.u.)能级差在所有体系中较高,使电子跃迁几率降低,预示其反应活性最低,最稳定,而iv3和iv4(0.31803a.u.)能级差相同且最小,故最不稳定的化合物可能是iv3和iv4。

图3生成焓与氮原子数的关系
Fig.3The relationship of the ΔHfand N atom numbers
表31,2,4,5-四嗪衍生物的EHOMO,ELUMO和ELUMO-HOMO

Table 3 Calculated HOMO and LUMO energies and energy gap (ΔELUMO-HOMO) of 1,2,4,5-tetrazine derivatives a.u.
3.4 爆轰性能
爆速和爆压是衡量含能材料爆轰性能的重要参数。本研究在wB97/6-31+G**水平下预测1,2,4,5-四嗪衍生物及RDX和HMX的爆速(D)和爆压(p),见表4。由表4可知,RDX和HMX的理论预测值与实验值对比较为接近,说明运用该方法得到的结果用于预测标题化合物的爆轰性能是相对可靠的。结果表明,带有不同取代基的1,2,4,5-四嗪衍生物密度不同,最大为1.84 g·cm-3,最小为1.60 g·cm-3,这也因此说明了目标化合物可能具备不同的D和p。通过比较i1和i2、ii1和ii2、iii1和iii2、iv1和iv2的ρ、D、p可以看出,四唑环上引入—NO2有利于增加1,2,4,5-四嗪衍生物的ρ、D、p; 比较i1和ii1,iii1和iv1,i2和i3的ρ、D、p可以看出,四唑环上引入—NN—使四嗪衍生物ρ减小,而D和p显著增加; 比较i1和iii1的ρ、D、p可以看出,四唑骨架能显著增加1,2,4,5-四嗪衍生物的ρ、D、p。取代基相同但位置不同的1,2,4,5-四嗪衍生物(i4和i5、i6和i7、i8和i9、ii4和ii5、ii6和ii7、ii8和ii9)的ρ、D、p计算结果不同但相近,说明取代基的位置不同对均四嗪衍生物的爆轰性能影响较小。
1,2,4,5-四嗪衍生物与RDX和HMX的ρ、D和p的比较如图4所示。从图4可以看出,有13种标题化合物的ρ接近RDX[44](1.82 g·cm-3),最高达到1.84 g·cm-3。D和p的趋势一致,有16种1,2,4,5-四嗪衍生物的D和p比RDX[44](8.75 km·s-1和34.00 GPa)高,且有12种化合物有较理想的爆轰性能,ρ、D和p分别接近于1.90 g·cm-3、9.00 km·s-1和40.00 GPa,其D超过HMX(9.10 km·s-1[44]),p接近于HMX(39 GPa[44]),尤其是iv3,D和p达到9.307 km·s-1,38.673 GPa,具有很好的爆轰性能,因此可以作为潜在的HEDM候选物。

图4目标化合物及RDX和HMX的ρ、D、p
Fig.4Calculatedρ,D, andpvalues of the title compounds together with RDX and HMX
表41,2,4,5-四嗪衍生物及RDX和HMX的平均摩尔体积(V)、摩尔质量(M)、理论密度(ρ)、爆热(Q)、爆速(D)和爆压(p)
Table4Predicted average molar volumes(V), molar mass(M), theoretical densities (ρ), heats of detonation (Q), detonation velocities (D) and detonation pressures (p) of 1,2,4,5-tetrazine derivatives together with RDX and HMX

compd.V/cm3·mol-1M/g·mol-1ρ/g·cm-3Q/J·g-1D/km·s-1p/GPai193.498150.0401.601466.2067.6924.45i2130.365240.0101.841825.0049.1437.55i3163.096296.0231.821911.4019.2738.32i4147.310268.0171.821878.7869.1937.77i5148.941268.0171.801865.6159.1136.82i6110.860195.0251.761721.2378.5431.91i7110.695195.0251.761701.9488.5131.73i8129.114223.0311.731800.7348.6332.27i9128.026223.0311.741768.1588.6332.35ii1109.196178.0461.631636.5368.1127.49ii2147.447268.0171.821906.0149.2237.96ii3183.323324.0291.771973.6699.2237.30ii4163.766296.0231.811940.9089.2938.40ii5163.353296.0231.811935.9279.2838.36ii6128.340223.0311.741835.0258.7132.95ii7127.936223.0311.741807.1648.6732.70ii8147.365251.0381.701872.2358.6932.34ii9144.572251.0381.741854.0898.8133.71iii1128.015218.0531.701449.8437.9727.22iii2170.421308.0231.811747.4968.8634.91iii3198.446364.0351.831829.7769.1637.66iii4183.604336.0291.831796.5209.0636.76iii5147.494263.0381.781642.3988.4831.69iii6164.581291.0441.771712.4078.6132.59iv1162.980274.0651.681674.6978.3529.68iv2200.528364.0351.821879.6009.1537.38iv3230.986420.0471.821933.1109.3138.67iv4216.398392.0411.811914.1229.2237.85iv5183.788319.0501.741806.1428.6932.83iv6200.288347.0561.731852.0738.7633.27RDX129.53222.041.71(1.82[44])1679.208.76(8.75[44])33.05(34.00[44])HMX162.87296.051.82(1.91[44])1672.409.11(9.10[44])37.08(39.00[44])
3.5 热稳定性
键级(BO)是衡量物质化学键强度的重要电子结构参数。键级越大,其间电子云密度重叠较多,表明该键越强,不易断裂。键离解能(EBD)为判断含能化合物的稳定性提供了非常有用的信息。一般来说,断开一个键所需能量越小,键越弱,越易成为引发键,也就是说,化合物灵敏度较高,不稳定。因此,键离解能常被用来衡量含能化合物热稳定性的相对顺序。表5列出了标题化合物几个相对较弱键的Wiberg键级及其键离解能。
通过比较化合物1,2,4,5-四嗪(473.93 kJ·mol-1)、i1(463.57 kJ·mol-1)和iii1(462.98kJ·mol-1)最弱键的EBD可以看出,四唑环的引入使1,2,4,5-四嗪衍生物的稳定性降低,但影响较小。通过比较化合物i1(463.57 kJ·mol-1)和ii1(220.42 kJ·mol-1)及化合物iii1(462.98 kJ·mol-1)和iv1(216.45 kJ·mol-1)最弱键的EBD可以看出,连接桥—NN—的加入使EBD降低约240 kJ·mol-1。通过比较化合物i系列中i1(463.57 kJ·mol-1)、i7(256.89 kJ·mol-1)和i2(252.03 kJ·mol-1)及iii系列中iii1(462.98 kJ·mol-1)、iii5(273.10 kJ·mol-1)和iii2(268.45 kJ·mol-1)最弱键的EBD可以看出,四唑环上引入—NO2使BDE降低约189~207 kJ·mol-1,但化合物i7和i2及iii5和iii2的EBD非常接近,相差约4 kJ·mol-1,表明随着—NO2数目的增多,四嗪衍生物的稳定性并无较大的变化。综上所述,四唑环与四嗪环直接连接使1,2,4,5-四嗪衍生物稳定性降低,但影响较小,而通过—NN—连接两者则明显地降低了其稳定性; 四唑环上引入—NO2或—NN—NO2使1,2,4,5-四嗪衍生物稳定性降低,且增加取代基数量对其稳定性影响较小; Rg1—Rg2、Rg1—R、Rg2—R或Rg2—NN(Rg1代表四嗪环,Rg2代表四唑环,R代表取代基)均有可能成为1,2,4,5-四嗪衍生物的热引发键。
表5目标化合物相对较弱键的Wiberg键级(BO, a.u.)和键离解能(EBD, kJ·mol-1)
Table5BO andEBDof the relatively weak bonds of the title compounds

compd.Rg1—Rg2BOEBDRg1—RBOEBDRg1—NNBOEBDRg2—RBOEBDRg2—NNBOEBDN—NO2(Rg1)BOEBDN—NO2(Rg2)BOEBDi10.99463.570.89482.680.89514.55i20.93453.340.86252.030.90269.12i30.98429.511.0024.261.0225.670.8448.430.7735.02i40.99427.790.86254.411.0225.340.7735.05i50.93456.201.0023.030.90270.600.8448.05i60.92460.100.89482.750.90273.02i71.01461.730.86256.890.89515.58i80.97432.850.89483.421.0227.530.7735.23i91.00462.781.0026.060.89515.240.8449.30ii10.90480.601.01317.090.89513.331.07220.42ii20.86253.941.01313.000.88260.231.0619.87ii31.0024.581.02315.601.0219.871.07175.970.8448.780.7426.85ii40.86266.041.02325.191.0224.051.08186.650.7637.55ii51.0023.931.01314.860.88267.501.05205.370.8348.48ii60.89481.471.00316.580.88262.621.04207.78ii70.86255.901.02316.350.89513.781.09220.07ii80.89506.031.01341.231.0438.901.05202.290.8351.47ii91.0025.231.02317.540.89520.381.09220.180.8449.21iii11.00462.980.89515.37iii20.93453.250.90268.45iii30.95443.551.0441.370.8251.67iii40.92428.181.0225.140.7735.30iii51.01458.770.90273.10iii61.00429.691.0225.780.7733.97iv11.02311.790.89516.491.08216.45iv21.01314.540.88260.911.05205.32iv31.02315.551.0220.051.06176.080.7427.09iv41.01294.461.0214.421.07177.070.7426.59iv51.01316.260.88268.351.05206.38iv61.02294.771.0220.401.07176.670.7427.26
Note: Rg1, Rg2and R represent the tetrazine ring, tetrazole ring and substituents,respectively.
图5比较了1,2,4,5-四嗪衍生物最弱键的BO和EBD的变化趋势。从图5可以看出,EBD和BO的变化趋势并不是完全吻合的,有的键级较小的化合物键离解能反而大,如化合物i2、i6、i7、iii2和iii5。所以,衡量一个物质的稳定性不但要看其BO,还要考虑其EBD的大小。Chung[45]等提出,高能量密度化合物最弱键的EBD高于80 kJ·mol-1,从图中可以看出,低于80 kJ·mol-1的1,2,4,5-四嗪衍生物EBD很小,在14~42 kJ·mol-1之间,稳定性不好,有14种1,2,4,5-四嗪衍生物的EBD高于80 kJ·mol-1,综合生成焓和爆轰性能的结果,化合物i2、ii2和iv2可以作为潜在的HEDM候选物。
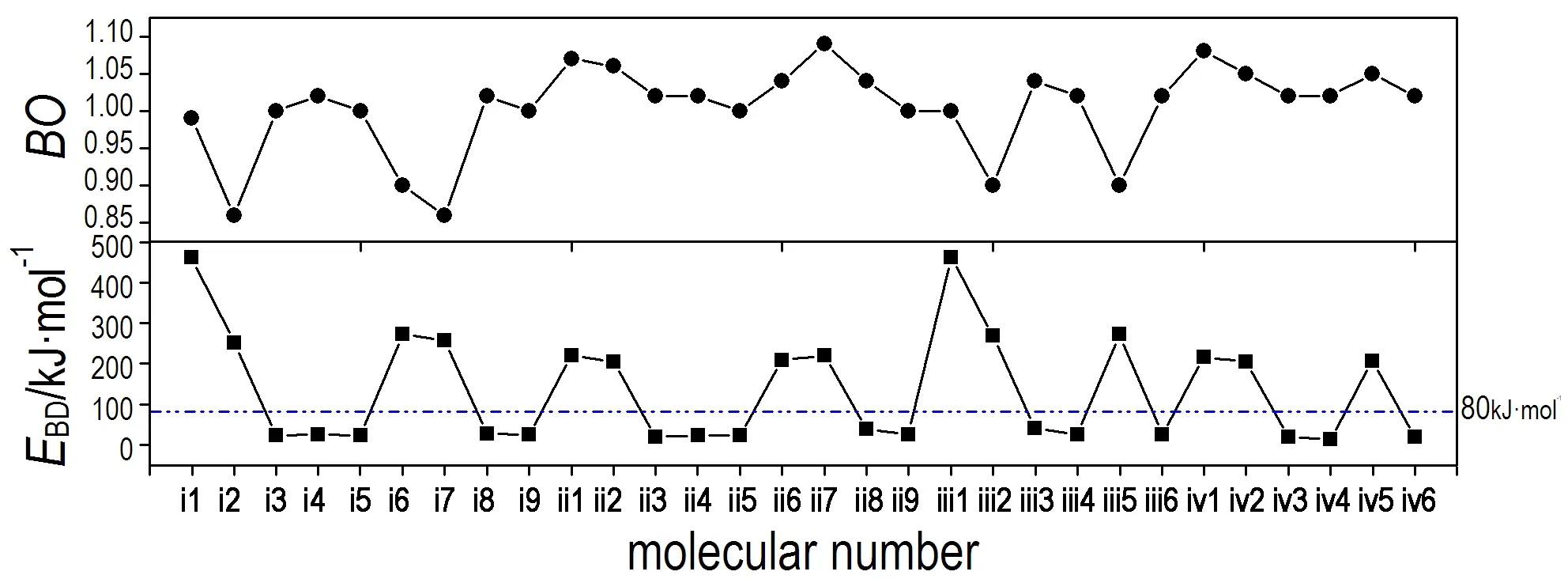
图5目标化合物较弱键的BO与EBD的比较
Fig.5The comparison ofBOandEBDof the relatively weak bonds of the title compounds
3.6 热力学性质
基于统计热力学,运用Gaussian09和自编程序,求得部分1,2,4,5-四嗪衍生物在200~800 K温度范围的热力学性质,即标准摩尔热容(cp)、标准摩尔熵(Sm)和标准摩尔焓(Hm)。通过线性拟合求得各目标化合物在200~800 K温度范围的cp、Sm和Hm与温度间关系如图6所示。由图6可见,cp及Hm均随温度的升高而增加,这主要是因为在较低温度时,分子的平动和转动对热力学函数的贡献大; 但是温度升高后,分子的振动增强,对热力学函数贡献大,从而导致热力学函数值增加。计算结果表明,在同一温度下,—NO2、—NN—及四唑骨架均有利于cp、Sm和Hm的增加,且对称双取代1,2,4,5-四嗪衍生物的比相应单取代的高。由图6还可以看出,标题化合物的cp和Sm的增幅均随温度的升高而逐步减小,而Hm的增幅均随温度的升高而逐步增大。通过iv2和iv3的热力学性质与温度间的函数关系式可以看出,曲线二次方项的系数非常小,近似为直线,化合物iv2和iv3的三个热力学函数值随温度的升高基本呈线性递增。其余化合物的热力学性质与温度间也存在类似的线性关系。

a.b.c.

d.e.f.
图6目标化合物cp、Sm和Hm与T的关系
Fig.6The relationships betweenTandcp、SmandHmof the title compounds
4 结 论
利用wB97/6-31+G**方法对四唑类1,2,4,5-四嗪衍生物的几何构型、前线轨道能量、生成焓、热稳定性、爆轰性能及热力学性质进行计算研究。计算结果表明,对称双取代对于提高1,2,4,5-四嗪的生成焓具有显著效果,同时含能骨架四唑环及在其上引入—NO2和—NN—NO2能够显著增加四嗪衍生物的生成焓; 爆轰性能结果表明,在四唑环上引入—NO2和—NN—能够显著提高1,2,4,5-四嗪衍生物的爆轰性能,且对称双取代对于提高衍生物的爆轰性能具有很好的效果; 通过分析标题化合物最弱键的键离解能,有14种化合物的EBD高于80 kJ·mol-1,具有较理想的热稳定性,在四唑环上引入—NO2和—NN—不利于提高衍生物的稳定性; 热容cp、熵Sm及焓Hm均随温度的升高而增加,且所有取代基均使cp、Sm、Hm值增大,cp和Sm的增幅随温度升高而逐步减小,而Hm的增幅则随温度的升高而逐步增大。综合分析,化合物i2、ii2和iv2从能量、爆轰性能和热稳定性上可以作为备选的高能量密度材料。
参考文献:
[1] Hiskey M A, Chavez D E, Naud D. Progress in high-nitrogen chemistry[C]∥In explosives, propellants and pyrotechnics. Proceedings of 27th International Pyrotechnics Seminar. July 16-21, USA, Colorado, 2000:3-14.
[2] 张兴高,朱慧,张炜,等. 高氮化合物在含能材料中的应用研究进展[J]. 含能材料, 2004, 12(Suppl.): 48-53.
ZHANG Xing-gao, ZHU Hui, ZHANG Wei, et al.Application development of poly-nitrogen compounds in energetic materials[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2004, 12(Suppl.): 48-53.
[3] 赵振国, 孙娜波, 饶国武. s-四嗪类化合物的研究进展[J]. 浙江化工, 2014, 45(1): 15-19.
ZHAO Zhen-guo, SUN Na-bo, RAO Guo-wu. Research advances in s-tetrazine compounds[J].ZhejiangChemicalIndustry, 2014, 45(1): 15-19.
[4] 熊鹰,舒远杰,王新锋,等. 四嗪类高氮化合物结构对热分解机理影响的理论研究[J]. 火炸药学报, 2008, 31(1): 1-5.
XIONG Ying, SHU Yuan-jie, WANG Xin-feng, et al. Theoretical study on effect of tetrazine structures on their thermal decomposition mechanisms[J].ChineseJournalofExplosives&Propellants, 2008, 31(1): 1-5.
[5] 徐松林,阳世清,王云鹏. 四嗪类高氮含能材料研究进展[J]. 化学推进剂与高分子材料, 2007, 5(1): 14-19.
XU Song-lin, YANG Shi-qing, WANG Yun-peng. Research advances in high-nitrogen energetic materials derived from tetrazine[J].ChemicalPropellants&PolymericMaterials, 2007, 5(1): 14-19.
[6] 周阳,龙新平,王欣,等. 高氮含能化合物的研究新进展[J]. 含能材料, 2006, 14(4): 315-320.
ZHOU Yang, LONG Xin-ping, WANG Xin, et al. Review on high-nitrogen energetic materials[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2006, 14(4): 315-320.
[7] 雷永鹏,徐松林,阳世清. 高氮含能化合物应用研究新进展[J]. 化学推进剂与高分子材料, 2007, 5(3): 1-14.
LEI Yong-peng, XU Song-lin, YANG Shi-qing. New research progress in application of high-nitrogen energetic compound[J].ChemicalPropellants&PolymericMaterials, 2007, 5(3): 1-14.
[8] 冯金玲, 张建国, 王昆, 等. 3,6-二肼基-1,2,4,5-四嗪的晶体结构及理论研究[J]. 高等学校化学学报, 2011, 32(7): 1519-1525.
FENG Jin-ling, ZHANG Jian-guo, WANG Kun, et al. Crystal structure and theoretical studies of 3,6-dihydrazino-1,2,4,5-tetrazine[J].ChemicalJournalofChineseUniversities, 2011, 32(7): 1519-1525.
[9] Qiong Wu, Yong Pan, Xuelan Xia, et al. Theoretic design of 1,2,3,4-tetrazine-1,3-dioxide-based high-energy density compounds with oxygen balance close to zero[J].StructChem, 2013, 24: 1579-1590.
[10] 徐松林, 阳世清, 张炜, 等. s-四嗪类高氮含能化合物的合成及性能[J]. 国防科技大学学报, 2006, 28(6): 17-23.
XU Song-lin, YANG Shi-qing, ZHANG Wei, et al. The synthesis and performance of s-tetrazine based high-nitrogen energetic compounds[J].JournalofNationalUniversityofDefenseTechnology, 2006, 28(6): 17-23.
[11] 潘劼, 何金选, 陶永杰. 3,6-二氨基-1,2,4,5-四嗪的合成及表征研究[J]. 含能材料, 2004, 12(增刊): 58-59.
PAN Jie, HE Jin-xuan, TAO Yong-jie. Synthesis and characterization of 3,6-diamine-1,2,4,5-tetrazine[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2004, 12(Suppl.): 58-59.
[12] 胡银,马海霞,张教强,等. 3,6-二氨基-1,2,4,5-四嗪二聚体分子间相互作用的理论研究[J]. 化学通报, 2010 (3): 263-268.
HU Yin, MA Hai-xia, ZHANG Jiao-qiang, et al. Theoretical study on intermolecular interactions of 3,6-diamino-1,2,4,5-tetrazine dimers[J].JournalofChemistry, 2010 (3): 263-268.
[13] 何冬梅,程广斌,吕春绪. 四嗪类高氮含能化合物的合成与表征[J]. 火炸药学报, 2010, 33(5): 8-11.
HE Dong-mei, CHENG Guang-bin, Lü Chun-xu. Synthesis and characterization of high-nitrogen energetic compounds derived from tetrazine[J].ChineseJournalofExplosives&Propellants, 2010, 33(5): 8-11.
[14] Huynh M H V, Hiskey M A, Archuleta J G, et al. 3,6-Di(azido)-1,2,4,5-tetrazine: a precursor for the preparation of carbon nanospheres and nitrogen-rich carbon nanospheres and nitrogen-rich carbon nitrides[J].AngewChemIntEd,2001, 43: 5658-5661.
[15] 阳世清,徐松林. 3,6-二氨基-1,2,4,5-四嗪-1,4-二氧化物的合成与表征[J]. 含能材料, 2005, 13(6): 362-365.
YANG Shi-qing, XU Song-lin. Synthesis and characterization of 3,6-diamino-1,2,4,5-tetrazine-1,4-dioxide[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2005, 13(6): 362-365.
[16] 徐松林, 阳世清, 岳守体, 等. 3,3′-偶氮-(6-氨基-1,2,4,5-四嗪)的合成与表征[J]. 合成化学, 2005, 13(6): 584-613.
XU Song-lin, YANG Shi-qing, YUE Shou-ti, et al. Synthesis and characterization of 3,3′-azobis(6-amino-1,2,4,5-tetrazine)[J].ChineseJournalofSynthesisChemistry, 2005, 13(6): 584-613.
[17] 张海昊,贾思媛, 王伯周, 等. 3,6-二肼基-1,2,4,5-四嗪及其含能盐的合成与性能[J]. 火炸药学报, 2014, 37(2): 23-30.
ZHANG Hai-hao, JIA Si-yuan, WANG Bo-zhou, et al. Synthesis and properties of 3,6-dihydrazine 1,2,4,5-tetrazine and its energetic salts[J].ChineseJournalofExplosives&Propellants, 2014, 37(2): 23-30.
[18] 岳守体, 阳世清. 3,6-双(1氢-1,2,3,4-四唑-5-氨基)-1,2,4,5-四嗪的合成与表征[J]. 合成化学, 2004, 12(2): 164-166.
YUE Shou-ti, YANG Shi-qing. Synthesis and characterization of 3,6-bis(1H-1,2,3,4-tetrazol-5-yl-amino)-1,2,4,5-tetrazine[J].ChineseJournalofSyntheticChemistry, 2004, 12(2): 164-166.
[19] 霍欢,王伯周,罗义芬,等. 3,6-双硝基胍基-1,2,4,5-四嗪(DNGTz)及其衍生物的合成、表征及热性能[J]. 固体火箭技术, 2013, 36(4): 500-505.
HUO Huan, WANG Bo-zhou, LUO Yi-fen, et al. Synthesis, characterization and thermal properties of energetic compound 3,6-dinitroguanidino-1,2,4,5-tetrazine (DNGTz) and its derivatives[J].JournalofSolidRocketTechnology, 2013, 36(4): 500-505.
[20] 孙谋, 张建国, 冯金玲, 等. 3,6-二(3,5-二甲基吡唑-1-基)-1,2,4,5-四嗪的晶体结构[J]. 含能材料, 2012, 20(6): 812-813.
SUN Mou, ZHANG Jian-guo, FENG Jin-ling, et al. Crystal structure of 3,6-bis(3,5-dimethylpyrazol-1-yl)-1,2,4,5-tetrazine[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2012, 20(6): 812-813.
[21] 肖鹤鸣, 陈兆旭. 四唑化学的现代理论[M]. 北京: 科学出版社, 2000: 1-266.
XIAO He-ming, CHEN Zhao-xu. The modern theory of tetrazole chemistry[M]. Beijing: Science Press, 2000, 1-266.
[22] 代玲玲, 崔胜峰, Damu G L V, 等. 四唑类化合物的合成及应用研究新进展[J]. 有机化学, 2013, 33: 224-244.
DAI Ling-ling, CUI Sheng-feng, Damu G L V, et al. Recent advances in the synthesis and application of tetrazoles[J].ChineseJournalofOrganicChemistry, 2013, 33: 224-244.
[23] 徐松林, 阳世清, 岳守体. 偶氮四唑类高氮含能化合物的合成及表征[J]. 火炸药学报, 2005, 28(3): 52-54.
XU Song-lin, YANG Shi-qing, YUE Shou-ti. Synthesis and characterization of high-nitrogen energetic compounds derived from azotetrazolate[J].ChineseJournalofExplosives&Propellants, 2005, 28(3): 52-54.
[24] Zhichao LIU, Qiong WU, Weihua ZHU, et al. Theoretical study of energetic trinitromethyl-substituted tetrazole and tetrazine derivatives[J].JPhysOrgChem, 2013, 26: 939-947.
[25] Qiong Wu, Weihua Zhu, Heming Xiao. Molecular design of tetrazole- and tetrazine-based high-density energy compounds with oxygen balance equal to zero[J].JChemEngData, 2013, 58: 2748-2762.
[26] 陈德霞. 四唑类含能化合物的合成及性能研究[D]. 硕士论文. 南京: 南京理工大学, 2012.
CHEN De-xia. The synthesis and properties of tetrazole energetic compounds[D]. M.D.Dissertation. Nanjing: Nanjing University of Science & Technology, 2012.
[27] Tao Yue, Mei-Xiang Wang, De-Xian Wang, et al. Asymmetric synthesis of 5-(1-hydroxyalkyl) tetrazoles by catalytic enantioselective passerini-type reactions[J].AngewChemIntEd, 2008, 47: 9454-9457.
[28] 邱丽美, 贡雪东, 郑剑, 等. 由原子化反应法求算高能化合物的生成热[J]. 含能材料, 2008, 16(6): 647-668.
QIU Li-mei, GONG Xue-dong, ZHENG Jian, et al. Heats of formation for energetic compounds calculated using atomization reactions[J].ChineseJournalofEnergeticMaterials, 2008, 16(6): 647-668.
[29] Yang Zhou, XinPing Long, YuanJie Shu. Theoretical studies on the heats of formation, densities, and detonation properties of substituted s-tetrazine compounds[J].JMolModel, 2010, 16: 1021-1027.
[30] Yong Pan, Weihua Zhu, Heming Xiao. Theoretical studies on the structures, heats of formation, energetic properties and pyrolysis mechanisms of nitrogen-rich difurazano[3,4-b:3′,4′-e]piperazine derivatives and their analogues[J].StructChem, 2013, 24: 1071-1087.
[31] Fang Wang, Guixiang Wang, Hongchen Du, et al. Theoretical studies on the heats of formation, detonation properties, and pyrolysis mechanisms of energetic cyclic nitramines[J].JPhysChemA, 2011, 115: 13858-13864.
[32] Xiao-hong Li, Rui-zhou Zhang, Xian-zhou Zhang. Theoretical studies of 1,2,4,5-tetrazine-based energetic nitrogen-rich compounds[J].StructChem, 2013, 24: 393-400.
[33] Kamlet M J, Jacobs S J. Chemistry of detonations. I. A simple method for calculating detonation properties of C—H—N—O explosives[J].JournalofChemicalPhysics, 1968, 48: 23-25.
[34] 王桂香, 肖鹤鸣, 居学海, 等. 含能材料的密度、爆速、爆压和静电感度的理论研究[J].化学学报, 2007, 65(6): 517-524.
WANG Gui-xiang, XIAO He-ming, JU Xue-hai, et al. Theoretical studies on densities, detonation velocities and pressures and electric spark sensitivities of energetic materials[J].ActaChimicaSinica, 2007, 65(6): 517-524.
[35] 李志敏, 严英俊, 冀慧莹, 等. 苦味酸含能离子盐的结构、生成热及爆炸性能理论研究[J]. 火炸药学报, 2009, 32(6): 6-10.
LI Zhi-min, YAN Ying-jun, JI Hui-ying, et al. Theoretical study on structure, heat of formation and detonation properties of picrate-based energetic ionic salts[J].ChineseJournalofExplosives&Propellants, 2009, 32(6): 6-10.
[36] 张熙和,云主慧. 爆炸化学[M]. 北京: 国防工业出版社, 1989: 1-344.
ZHANG Xi-he, YUN Zhu-hui. Explosion chemistry[M]. Beijing: National Defence Industry Press, 1989: 1-344.
[37] Benson S W. Thermochemical kinetics. 2nd ed.; New York: Wiley-Interscience, 1976.
[38] Mills I, Cvitas T, Homann K, Kallay N, et al. Quantities, units, and symbols in physical chemistry. Oxford: Blackwell Scientific Publications, 1988: 1-233.
[39] Blanksby S J, Ellison G B. Bond dissociation energies of organic molecules[J].AccChemRes, 2003, 36: 255263.
[40] 汤正新, 李小红, 张瑞州. 硝酸酯含能材料中O—N键离解能的计算[J]. 河南科技大学学报, 2012, 33(3): 80-85.
TANG Zheng-xin, LI Xiao-hong, ZHANG Rui-zhou. Calculation of bond dissociation energies of O—N bond for some nitroester energetic materials[J].JournalofHenanUniversityofScienceandTechnology, 2012, 33(3): 80-85.
[41] 李小红,牛芳. 取代氯苯化合物的C—Cl键离解能的密度泛函研究[J]. 新乡学院学报, 2009, 26(3): 40-44.
LI Xiao-hong, NIU Fang. DFT study of the C—Cl bond dissociation enthalpies of substituded chlorobenzene compounds[J].JournalofXinxiangUniversity, 2009, 26(3): 40-44.
[42] 邵菊香, 程新路, 杨向东, 等. 对一些叠氮化合物的叠氮自由基键离解能的计算[J]. 四川师范大学学报, 2007, 30(1): 78-82.
SHAO Ju-xiang, CHENG Xin-lu, YANG Xiang-dong, et al. The calculation of bond dissociation energies for azide group in Some azido compounds[J].JournalofSichuanNormalUniversity, 2007, 30(1): 78-82.
[43] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision A.02. Gaussian, Inc.: Wallingford, CT, 2009.
[44] Talawar M B, Sivabalan B, Mukundan T, et al. Environmentally compatible next generation of ′green′ energetic materials(GEMs)[J].JHazardMater, 2009, 161(2-3): 589-607.
[45] Chung G S, Schmidt M W, Gordon M S. An ab initio study of potential energy surfaces for N8 isomers[J].JPhyChemA, 2000, 104(23): 5647-5650.
猜你喜欢
心肺血管病杂志(2019年4期)2019-06-27 07:36:10
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
数学理论与应用(2016年4期)2016-05-17 04:50:26
小学生导刊(低年级)(2016年2期)2016-02-24 22:39:09
合成化学(2015年10期)2016-01-17 08:56:26
中国塑料(2015年11期)2015-10-14 01:14:05
应用数学与计算数学学报(2014年4期)2014-09-26 12:15:53
华东师范大学学报(自然科学版)(2014年1期)2014-04-16 02:54:58
火炸药学报(2014年5期)2014-03-20 13:17:51
火炸药学报(2014年5期)2014-03-20 13:17:46
