
α-取代苯并噻吩钌乙炔及钌乙烯配合物的合成、表征、电化学及光谱电化学性质
2016-05-03 07:06欧亚平张静邝代治张复兴庾江喜朱小明
无机化学学报 2016年4期
欧亚平张 静邝代治张复兴庾江喜朱小明
(1衡阳师范学院化学与材料科学学院,功能金属有机材料湖南省普通高等学校重点实验室,衡阳 421008) (2华中师范大学化学学院,农药与化学生物学教育部重点实验室,武汉 430079)
α-取代苯并噻吩钌乙炔及钌乙烯配合物的合成、表征、电化学及光谱电化学性质
欧亚平*,1张静2邝代治1张复兴1庾江喜1朱小明1
(1衡阳师范学院化学与材料科学学院,功能金属有机材料湖南省普通高等学校重点实验室,衡阳421008) (2华中师范大学化学学院,农药与化学生物学教育部重点实验室,武汉430079)
摘要:以α-取代苯并噻吩乙炔及乙烯基为配体,与不同辅助配体键合的金属钌为氧化还原活性端基,分别合成并表征了α-取代苯并噻吩钌乙炔及钌乙烯配合物1和2,两个配合物均经X射线单晶衍射的确证。电化学性质表明配合物1具有较好的氧化还原可逆性,金属氧化态相对于2比较稳定。红外光谱电化学研究表明配合物1和2分别形成1+和2+后,与金属相连的ν(C≡C)和ν(C≡O)伸缩振动吸收均发生变化,其中ν(C≡C)变化较大,可能由于形成了Ru=C=C结构;紫外可见近红外光谱电化学研究发现电解后,中性分子1和2在紫外区域的强吸收峰均逐渐降低,1+分别在可见及近红外580 nm,698 nm及874 nm处出现较弱的3个吸收峰,3个吸收峰可能归属于LMCT吸收。2+在296 nm处出现1个很弱的吸收峰,这不同于配合物1+,可能源于2+的稳定性。
关键词:α-取代苯并噻吩;钌乙炔配合物;钌乙烯配合物;合成;电化学;光谱电化学
功能金属有机材料湖南省普通高校重点实验室开放基金(No.GN15K05)和衡阳师范学院科学基金(No.14B23)资助项目。*通信联系人。E-mail:ouyaping123@126.com
0引 言
近年来,电子器件的微型化促使基于分子水平上的电子器件的研究日益显著。分子电子器件按其功能不同也分为分子导线、分子开关、分子整流器及分子存储器[1-3]。分子导线作为分子电子器件中的桥梁及关键单元已经引起了研究者们的广泛关注[4-6]。其中,共轭配体桥链的双核金属配合物可以作为研究分子导线的模型分子,其合成及电子性质研究已经成为科学家们的重要研究课题和前沿领域[7-8]。
作为研究分子导线的模型分子,双核金属配合物中必须存在2个关键单元:其一是研究主体-共轭桥链配体;其二是氧化还原活性金属端基(电化学活性),这样就能在电化学作用下使分子形成电子给体-共轭桥链配体-电子受体,由于共轭桥链配体的电子传递作用,通过电化学测试就能够判断桥配体作为分子导线的电子传递能力[7,9]。配合物的电子性质与共轭金属本身的性质及配体的性质有关[10-11]。为了考察不同配体下同种金属的电子性质,本文选择富电子性且易修饰的苯并噻吩为配体,不同辅助配体的金属钌为氧化还原活性端基,分别合成及表征了2种α-取代苯并噻吩钌乙炔及钌乙烯单核配合物,并通过光谱及电化学技术研究2种配合物的电子性质。
1 实验部分
1.1试剂与仪器
实验过程中大部分反应都是在氮气或者氩气保护下进行的,采用的是标准的Schlenk双排管真空线技术。溶剂四氢呋喃经钠/二苯甲酮处理,二氯甲烷和正己烷用氢化钙处理,甲苯经钠/二苯甲酮处理,三乙胺用KOH干燥处理,使用前将处理好的溶剂新蒸出并脱气。其他常用溶剂从中国医药上海化学试剂公司购得后直接使用。试剂正丁基锂、三甲基硅乙炔(TMSA)、苯并噻吩直接从武汉格奥试剂公司购买得到,另外一些起始原料RuCl(dppe)Cp*[12]、RuHCl(CO)(PPh3)3[13]根据文献方法合成得到。
元素分析(C、H、N)经华中师大分析测试中心Vario ElⅢCHNSO元素分析仪测得。1H NMR、13C NMR和31P NMR经Varian MERCURY Plus 400 MHz或Varian MERCURY Plus 600 MHz核磁共振仪测得。其中1H NMR、13C NMR谱化学位移以TMS作为内标,31P NMR谱化学位移以85% H3PO4作为外标。
化合物的单晶衍射数据均在Bruker Apex CCD X射线衍射仪下收集,其晶体衍射数据经过SAINT v6.26还原[14]。晶体结构用SHELXTL-97[15]程序中的直接法解出,部分非氢原子坐标在随后的数轮差值傅立叶合成中确定。对所有非氢原子坐标及其各向异性热参数进行了全矩阵最小二乘法修正,氢原子用理论加氢的方式产生。
CCDC:1441747,1;1443680,2。
液体红外光谱均使用AVATAR 360型FT-IR红外光谱仪(液体红外池)测定,紫外可见近红外光谱使用日本岛津公司生产的UV-3600紫外-可见-近红外光谱仪测定。
电化学测量采用的是电化学工作站CHI 660C (CH Instruments Company,USA)。以玻碳电极作为工作电极,铂电极作对电极,Ag+|Ag电极为参比电极。以0.001 mol·L-1n-Bu4NPF6的CH2Cl2溶液为电解液,被测底物的浓度0.001 mol·L-1。循环伏安通常以扫描速率为100 mV·s-1测得,方波伏安在f=10 Hz条件下测得。光谱电化学通过Hartl教授设计的薄层电解池(OTTLE)[16]与电化学工作站联用测得(以CH2Cl2作为溶剂,0.001 mol·L-1n-Bu4NPF6作为支持电解质)。
光谱电化学实验在室温薄层电解池(OTTLE)中进行,该电解池内部结构为:一个铂网工作电极,路径长为0.2 mm的氟化钙窗口。将池子放在FT-IR红外光谱仪或者UV-3600紫外-可见-近红外光谱仪中,通过CHI 600C工作站进行控制电位电解实验。
1.2化合物的合成
目标化合物1和2的合成路线如图所示:

图1 目标化合物1及2的合成路线Fig.1 Synthetic paths to target complexes 1 and 2
1.2.1中间体1b及1c的合成
1b:安装好带有恒压滴液漏斗的100 mL三颈瓶装置,抽充氮气3次,向体系中加入乙醚(25 mL),苯并噻吩(7.45 mmol,1.0 g)。最后往体系中加入正丁基锂(8.20 mmol,3.28 mL)。体系在室温下搅拌3 h后,从恒压滴液漏斗中加入液溴(8.20 mmol,1.312 g),体系在冰浴下反应10 min,再继续反应2h后,用5%的氢氧化钾溶液猝灭反应,饱和食盐水洗涤,无水硫酸钠干燥,经柱色谱(石油醚作为洗脱剂)得到1.2 g白色固体,产率:76%。1H NMR (400 MHz,CDCl3):δ 7.27 (s,1H,thiophene),7.28 ~7.26 (br,2H,Ar),7.62 (d,J (HH) =6.8 Hz,1H),7.66 (d,J (HH) =7.6 Hz,1H)。
1c:在100 mL的二颈烧瓶中加入2-溴苯并噻吩1b(1.416 mmol,300 mg),Pd(PPh3)4(0.14 mmol,164 mg),CuI(0.14 mmol,27 mg),体系抽充氮气3次后,向其中加入脱气的THF溶液(20 mL)及三乙胺(20 mL)。室温下加入三甲基硅乙炔(3.54 mmol,0.35 g),然后体系加热回流24 h。冷却到室温,二氯甲烷溶解,饱和氯化钠溶液洗涤,合并有机相,无水硫酸钠干燥,经柱色谱(石油醚作为洗脱剂)得到黄色油状液体180 mg,产率:55%。1H NMR(400 MHz,CDCl3):δ 0.30(s,9H,SiMe3),7.34~7.37(br,2H,Ar),7.74(t,J(HH) =9.2 Hz,2H)。
1.2.2目标化合物1及2的合成
目标化合物1:在100 mL的二颈烧瓶中加入化合物1c (0.43 mmol,100 mg),RuCp*(dppe)Cl (0.43 mmol,288 mg),KF(2.58 mmol,150 mg),抽充氮气3次,向体系中注入甲醇(20 mL)及THF(5 mL)。体系加热回流24 h后,抽滤,得到的固体用二氯甲烷/正己烷重结晶得到170 mg黄色针状固体1。产率:49%。1HNMR(400 MHz,CDCl3):δ1.60(s,15H,C5(CH3)5), 2.07(br,2H,CH2/dppe),2.68(br,2H,CH2/dppe),6.48(s,1H,thiophene),7.10(t,J(HH)=7.2 Hz,1H),7.17(t,J(HH)= 7.6 Hz,1H),7.19~7.38(m,16H,HAr/dppe),7.46(d,J(HH) =3.6 Hz,1H),7.53 (d,J(HH)=4.4 Hz,1H),7.70~7.80 (m,4H,HAr/dppe)。13C NMR (100 MHz,CDCl3):δ 9.97 (CH3),29.38(CH2),92.89(C5H5),102.44(thiophene-C≡C),119.41(Ru-C≡C),121.31,121.46,122.34,123.61,127.41,129.01,132.03,133.18,133.52,136.26,136.7 4,138.14,138.47,140.76,143.60。31P NMR(160 MHz,CDCl3):δ 78.01。元素分析按C47H47P2RuS计算值(%): C,69.92;H,5.90;实测值(%):C,69.95;H,5.87。
目标化合物2:氮气保护下,向含有RuHCl(CO) (PPh3)3(314 mg,0.33 mmol)的20 mL二氯甲烷溶液的100 mL三颈瓶中,搅拌下缓慢滴加含1d(35 mg,0.22 mmol)的5 mL二氯甲烷溶液,滴加完毕后,混合物在室温下搅拌1~2 h,此时溶液变为红棕色。再向体系中加入PMe3(1 mol·L-1THF溶液)(2.20 mL,2.20 mmol),混合物继续在室温下反应20 h后,减压浓缩溶液至2~3 mL,再向其中加入大约30 mL正己烷,析出黄色固体90 mg,产率:74%。1H NMR (400 MHz,CDCl3):δ 1.41(t,J(PH)=3.2 Hz,18H,PMe3),1.48(d,J(PH)=7.2 Hz,9H,PMe3),6.75(s,1H,thiophene-H),6.82~6.87(m,1H,thiophene-CH=),7.16(t,J(HH)= 6.8 Hz,1H),7.20(t,J(HH)=7.6 Hz,1H),7.59(d,J(HH) =8.0 Hz,1H),7.68(d,J(HH)=8.0 Hz,1H),8.07~8.14(m, 1H,Ru-CH=)。13C NMR(100 MHz,CDCl3):δ 16.48(t,J=15.3 Hz,PMe3),19.80 (d,J=21.3 Hz,PMe3),113.30 (thiophene-CH=),121.83,122.41,122.49,123.79,128.77, 137.93,141.13,149.65(Ru-CH=),202.0(CO)。31P NMR (160 MHz,CDCl3):δ-18.60(t,J=22.72 Hz,PMe3),-7.20 (d,J=22.56 Hz,PMe3)。IR(KBr,cm-1):ν(CO) 1 910(s),ν(C=C)1539(m)。元素分析按C20H35ClOP3RuS计算值(%):C,43.44;H,6.38。实测值(%):C,43.59;H,6.23。
2 结果与讨论
2.1配合物1和2的结构表征
配合物1和2均通过核磁、元素分析及X射线单晶衍射确证。在1H NMR中,配合物1是以RuCp*(dppe)为端基的钌乙炔配合物,显示出了类似金属配合物的特征质子信号[17],在δ=1.60为五甲基环戊二烯上甲基氢信号,在2.07及2.68处为dppe上2 个CH2的质子信号;对于配合物2,在1.41及1.48处出现2个CH3的质子信号,分别对应于1组双重峰和1组三重峰,从而表现出2个不同的化学环境。由于受到金属钌的影响,碳碳双键发生了明显的耦合裂分,特别是与金属钌相连的碳上的氢的化学位移明显移向低场。在13C NMR中,2个配合物也同样出现特征的碳核信号,此外配合物1和2的31P NMR有明显的区别,配合物1呈现出1个磷信号,且位于低场,而配合物2出现2个磷信号,位于高场,这主要是由于磷与金属钌存在2个不同的配位环境所致,也就是通常所讲的“子午”配位[18]。
适合X射线分析的配合物1和2的单晶分别通过将正己烷扩散到含样品的二氯甲烷溶液中获得。图2呈现了配合物1和2的单晶结构,表1和2分别列出了配合物1和2的晶体学数据(包括晶体数据、数据收集及精修参数)及部分键长及键角参数。从图中可以看出,配合物1呈现类似的假八面体的几何构型,C(37)-C(38)的键长为0.119 5 nm,符合正常的碳碳三键键长范围,金属钌与三键之间形成的是Ru-C σ键,且Ru(1)-C(37)键长为0.200 8 nm,明显长于普通碳碳单键的键长。从键角来看,尽管RuCp*(dppe)的空间位阻较大,C(38)-C(37)-Ru(1)的键角仍为178.79°以及C(37)-C(38)-C(39)的键角为178.13°,说明受空间效应的影响较少。配合物2的晶体结构及数据显示,苯并噻吩与钌乙烯键基本处在同一平面上,钌氢配体与末端炔反应是以反式插入进行的,从而得到如图所示的反式双键。

图2 配合物1和2的分子结构椭球图Fig.2 Molecular structures of complexes 1 and 2
2.2电化学性质研究
为了理解2个配合物的电化学性质,我们采用循环伏安(CV)及方波伏安法(SWV)表征了配合物1 及2的电化学行为,对应的循环伏安及方波伏安图如图3和图4所示,表3列出了1和2的电化学数据。从图中可以看出,2个配合物均发生了1次单电子的氧化还原过程,配合物1的氧化电位明显高于配合物2。此外,从氧化电位和还原电位的峰电流之比可以看出,配合物1的氧化还原可逆性明显较好,也表明配合物1在被氧化成+1价后形成的阳离子比较稳定。
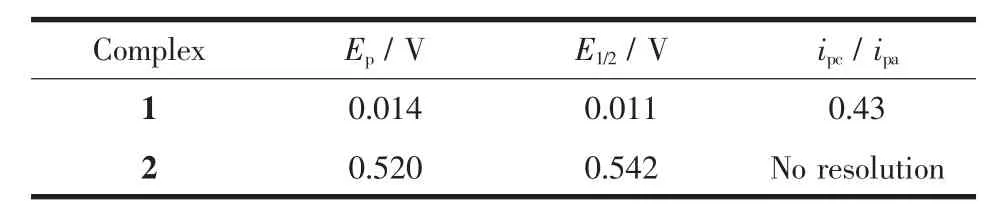
表3 配合物1及2的电化学数据aTable3 Electrochemical dataafor complexes 1 and 2

图3 配合物1的循环伏安(a)及方波伏安(b)曲线图Fig.3 Cyclic voltammograms (a) and square-wave voltammograms (b) of complex 1
2.3光谱电化学测试
2.3.1红外光谱电化学研究
红外光谱电化学能够很好地鉴别化合物在氧化前后结构中特征官能团的变化,如C≡C或者C≡O。配合物1中含有C≡C,配合物2中含有C≡O。那么通过电解实验,就能够原位检测这两种特征官能团伸缩振动吸收峰的变化。对应的红外吸收光谱变化(C≡C或者C≡O区域)如图5所示,不同价态下对应的ν(C≡C)及ν(C≡O)数据列于表4。从图中可以看出,配合物1和2在中性态下分别在2 049 cm-1(ν(C≡C))及1 923 cm-1(ν(C≡O))处出现1个很强的伸缩振动吸收峰。随着电解的进行,配合物由0价变为+1价,中性态下的吸收峰逐渐减弱。对于配合物1而言,被氧化之后,其在较低波数处1 972及1 880 cm-1出现2个较弱的吸收峰,这可能由于氧化之后钌炔配合物形成了Ru=C=C结构[19]。钌烯配合物2在电解之后,在较高波数处1 949及1 976 cm-1处出现了2个吸收峰,在1 976 cm-1的吸收峰可能是2+物种不稳定分解所产生的吸收,在1 949 cm-1的吸收峰是由于金属钌的氧化,导致与金属相连的C≡O伸缩振动吸收发生了一定的移动,且其变化相对于ν(C≡C)变化较小,表明氧化后形成的自由基阳离子可能定域在金属上的比例较少。

图4 配合物2的循环伏安(a)及方波伏安(b)曲线图Fig.4 Cyclic voltammograms (a) and square-wave voltammograms (b) of complex 2

图5 配合物1及2在薄层电解池(OTTLE)中一步单电子氧化过程的红外吸收光谱改变图: (a) 1→1+(ν(C≡C)区域); (b) 2→2 (ν(C≡O)区域)Fig.5 IR absorption spectra changes of single-electron oxidation for complexes 1 and 2 at OTTLE: (a) 1→1+(ν(C≡C) region); (b) 2→2+(ν(C≡O) region)
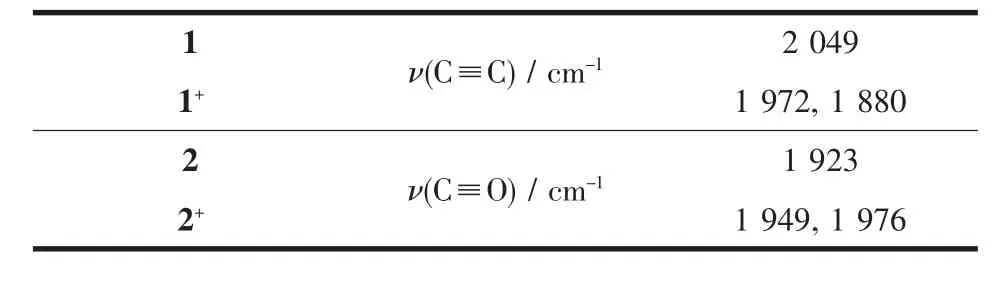
表4 红外吸收光谱变化(C≡C或者C≡O区域)数据Table4 Summary of the IR spectral data (ν(C≡C) or ν(C≡O)) for the complexes 1 and 2
2.3.2紫外可见近红外光谱电化学研究
紫外可见近红外光谱电化学原位检测能够跟踪不同价态价电子吸收光谱的变化情况。配合物1 及2在OTTLE中单电子氧化过程的紫外可见近红外吸收光谱变化如图6所示,中性态及单氧化态对应的电子吸收光谱数据列于表5。配合物1和2在中性态下的强吸收峰可能主要来自于共轭配体ππ*的跃迁及MLCT跃迁吸收[20]。配合物1在电解形成1+后,中性态在358 nm处的强吸收峰逐渐降低,同时分别在可见及近红外580、698及874 nm处出现较弱的3个吸收峰,这可能也是由于形成了Ru= C=C结构使得分子的共轭性增强,导致吸收峰出现在长波长方向,这3个吸收峰可能归属于LMCT吸收[19]。配合物2在电解后,紫外区域的强吸收峰减弱,同时在296 nm处出现1个很弱的吸收峰,在可见及近红外区域没有观察到吸收峰的变化,这不同于配合物1+,可能源于2+的稳定性。

图6 配合物1及2在薄层电解池(OTTLE)中一步单电子氧化过程的紫外可见近红外吸收光谱变化图Fig.6 UV-Vis-NIR absorption spectra changes of single-electron oxidation for complexes 1 and 2 at OTTLE

表5 不同氧化态1n+和2n+(n=0, 1)的电子吸收光谱数据Table5 Electronic absorption data of different single-oxidized states 1n+and 2n+(n=0, 1)
3结 论
本文合成了α-取代苯并噻吩钌乙炔及钌乙烯配合物1和2,并对其结构和其不同价态下的电子性质进行了表征,2个配合物的结构均经过了X射线单晶衍射的确证。电化学性质表明配合物1的氧化还原可逆性较好。对1和2的光谱电化学的研究,深入理解了它们在形成氧化态1+及2+的稳定性及电子跃迁情况,从而为后续的分子导线的研究模型双核金属钌配合物的研究提供了理论依据和对照。
参考文献:
[1] LI Rong-Jin(李荣金), LI Hong-Xiang(李洪祥), TANG Qing-Xin(汤庆鑫), et al. Physics(物理), 2006,35(12):1003-1009
[2]LI Yan-Wei(李延伟), ZHANG Zheng-Gang(张正刚), YAO Jin-Huan(姚金环), et al. Mater. Rev.(材料导报), 2009,23 (21):22-25
[3] Song H, Reed M A, Lee T. Adv. Mater., 2011,23:1583-1608
[4] YAO Chuan(姚川), LU Qi(路崎), WANG Xian-Hong(王献红), et al. Acta Polym. Sin.(高分子学报), 2014,0(12):1659-1668
[5] ZHANG Xiang-Yi(张相宜), ZHENG Qi(郑启), QIAN Chen-Xi(钱晨熹), et al. Chinese J. Inorg. Chem.(无机化学学报), 2012,28(8):1629-1634
[6] Frampton M J, Anderson H L. Angew. Chem. Int. Ed., 2007, 46:1028-1064
[7] HAN Mei-Juan(韩美娟). Chemistry(化学通报), 2012,75(6): 496-501
[8] Aguirre-Etcheverry P, O′Hare D. Chem. Rev., 2010,110: 4839 -4864
[9] WANG Wen-Zhen(王文珍), LIAO Dai-Zheng(廖代正), WANG Geng-Lin(王耕霖). Chemistry(化学通报), 2002,65 (2):107-112
[10]GONG Jian(龚剑), LI Guo-Ping(李国平). Chinese J. Inorg. Chem.(无机化学学报), 1995,11(3):107-112
[11]Costuas K, Rigaut S. Dalton Trans., 2011,40:5643-5658
[12]Bruce M I, Ellis B G, Low P J, et al. Organometallics, 2003, 22:3184-3198
[13]Ahmad N, Levison J J, Robinson S D, et al. Inorg. Synth., 1974,15:45-64
[14]SMART and SAINT, Bruker AXS Inc., Madison, 2002.
[15]Sheldrick G M. SHELXTL-97, Program for Crystal Structure Analysis, University of Göttingen, Germany, 1997.
[16]Krejcˇík M, Daněk M, Hartl F. J. Electroanal. Chem. Interfacial Electrochem., 1991,317:179-187
[17]Ou Y P, Xia J, Liu S H, et al. Chem. Asian J., 2013,8:2023 -2032
[18]Ou Y P, Jiang C, Liu S H, et al. Organometallics, 2011,30: 5763-5770
[19]Gao L B, Liu S H, Chen Z N, et al. Inorg. Chem., 2007,46: 5651-5664
[20]ZHANG Jing(张静), OU Ya-Ping(欧亚平), LIU Sheng-Hua(刘盛华), et al. Chin. Sci. Bull.(科学通报), 2014,17:1647-1654
Syntheses, Characterization, Electrochemistry and Spectroelectrochemistry of α-Benzothiophene Acetyl and Vinyl Mononuclear Ruthenium Complexes
OU Ya-Ping*,1ZHANG Jing2KUANG Dai-Zhi1ZHANG Fu-Xing1YU Jiang-Xi1ZHU Xiao-Ming1
(1College of Chemistry and Material Science, Hengyang Normal University; Key Laboratory of Functional Organometallic Materials of Hengyang Normal University, College of Hunan Province, Hengyang, Hunan 421008, China)
(2College of Chemistry, Central China Normal University; Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, Wuhan 430079, China)
Abstract:Using α-benzothiophene acetyl and vinyl as ligands, metal Ru with different auxiliary ligands as redoxactive terminus, we synthesized and characterized α-benzothiophene substituted mononuclear ruthenium acetyl and vinyl complexes 1 and 2. Two complexes were confirmed by X-ray single crystal diffraction. Electrochemical properties suggest that complex 1 has better redox reversibility, corresponding oxidation state 1+is more stable than 2+; IR Spectroelectrochemical studies found that 1 and 2 are oxidized into respective 1+and 2+, and respective ν(C≡C) and ν(C≡O) stretching vibration absorptions both have obvious changes: changes of ν(C≡C) is larger, which may be result from the formation of Ru =C =C; UV-Vis-NIR spectroelectrochemistry shows that after electrolytic, absorptions of neutral 1 and 2 in UV region fall down gradually, and 1+displayed three weak absorptions in visible and NIR region (580 nm, 698 nm and 874 nm), which may be assigned to LMCT contributions. 2+is different from 1+, which appears only a weak peak at 296 nm, and may be attributable to the stability of 2+. CCDC: 1441747, 1; 1443680, 2.
Keywords:α-substituted benzothiophene; ruthenium acetyl complex; ruthenium vinyl complex; synthesis; electrochemistry; spectroelectrochemistry
收稿日期:2015-12-12。收修改稿日期:2016-02-24。
DOI:10.11862/CJIC.2016.088
中图分类号:O614.82+1
文献标识码:A
文章编号:1001-4861(2016)04-0641-08
猜你喜欢
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
化工管理(2021年7期)2021-05-13
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
表面工程与再制造(2019年6期)2019-08-24
广东教育·高中(2018年12期)2018-02-13
资源节约与环保(2018年1期)2018-02-08
现代商贸工业(2016年14期)2016-12-27
安徽理工大学学报·自然科学版(2016年4期)2016-12-23
考试周刊(2016年85期)2016-11-11
农业与技术(2016年15期)2016-11-09
