
小花草玉梅正常和自然变异植株的AP3-3基因研究
2016-03-24 06:32刘虎岐
西北植物学报 2016年2期
张 婷,邢 妮,王 超,刘虎岐
(西北农林科技大学 生命科学学院,陕西杨陵 712100)
小花草玉梅正常和自然变异植株的AP3-3基因研究
张婷,邢妮,王超,刘虎岐*
(西北农林科技大学 生命科学学院,陕西杨陵 712100)
摘要:野生小花草玉梅(Anemone rivularis var.flore-minore)正常植株和花被片自然变异植株的外观形态差异很大,该研究以二者为材料,利用常规PCR和高效热不对称PCR(Hi-Tail PCR)技术从其正常和变异植株的基因组中各分离得到1个B类基因。序列分析证明,二者隶属于B类MADS-box基因AP3家族的旁系同源基因AP3-3分枝,分别命名为NArAP3-3(正常植株)和VArAP3-3(变异植株)。NArAP3-3基因全长3 795 bp,VArAP3-3基因全长3 898 bp,二者均含有1个666 bp的开放阅读框(ORF),可编码221个氨基酸,具有典型的植物MADS-box基因结构,其编码肽链包含了MADS区、K区、Ⅰ区和C区。对比NArAP3-3和VArAP3-3基因的全长序列,发现VArAP3-3基因比NArAP3-3多了1段49 bp的插入,且在ORF序列与NArAP3-3基因相比有4个碱基突变。对二者的全长序列、所编码的221个氨基酸及插入序列的生物信息学分析显示,二者在基因启动子、蛋白质基本性质、结构功能域、二级三级预测结构等方面均有差异,推测这些差异可能是花被片变异产生的原因之一。该研究结果为进一步探索其变异机制奠定了基础。
关键词:小花草玉梅;花被片变异;Hi-Tail PCR;MADS-box基因;NArAP3-3和VArAP3-3基因
花是被子植物独有的非常重要的观赏及生殖器官。植物花发育可分为成花诱导、花的发端、花器官发育及花型发育4个阶段,分别由花序分生组织特性基因、花分生组织特征基因[1-2]、同源异型基因[3]和背腹特性基因调控其表达。花器官发育的ABCDE[4-8]模型及“四因子[9-11]”模型可以很好地解释其形成过程中各同源异型基因的互作。“ABCDE[4-8]”模型是以核心真双子叶模式植物为基础逐步确立的,其花在形态上具有典型的萼片、花瓣、雄蕊和雌蕊四轮结构;而核心真双子叶植物的花器官发育模型在基部真双子叶植物中并不是严格保守的[12-13],基部真双子叶植物中B功能基因的表达区域可能扩展到外层导致花瓣状器官的分化,使外轮器官与内层花瓣在形态上具有一致性。ABCDE 基因中绝大多数都属于MADS-box基因[14-15],所以B类MADS-box基因在整个花器官发育过程中起着十分关键的作用。被子植物B类基因包含AP3和PI两个进化系,它们是由一个共同祖先基因复制而来的水平同源基因,编码的蛋白质之间可形成异二聚体,是多种植物花瓣和雄蕊发育所必需的,且经微阵列技术研究表明,AP3/PI直接调节花瓣和雄蕊细胞形成过程中基因的表达。AP3家族包括AP3-1、AP3-2和AP3-3等3个进化枝。目前关于AP3-3基因的研究较少且主要集中在毛茛科中。有研究表明,AP3-3基因通常只在花瓣中特异性表达且表达水平一般很高[16-17],在耧斗菜[17]和黑种草[17]中沉默AP3-3基因,仅造成其花瓣向萼片的同源异型转变而其他花器官大部分保持不变,这说明了AP3-3基因在控制花瓣形成过程中的关键作用。所以,AP3-3基因被称为花瓣身份特征基因,在植物花器官变化的研究中具有重要的价值和意义。
小花草玉梅(Anemonerivularisvar.flore-minore)属毛茛科银莲花属,是基部真双子叶植物,多年生草本,其正常花的花部集成二歧聚伞花序,花被无分化,由5(~8)枚花被片组成,雄、雌蕊多数[18]。基部真双子叶植物中B功能基因表达区域的外延现象在小花草玉梅花器官发育中有一定体现,它的花被无分化,花被片花瓣化,白色,行使花瓣的功能,这种在进化过程中形成的极似花瓣的花被片很可能也受花瓣特性基因AP3-3的影响。另Kramer研究[19-20]表明,该基因在毛茛科的多数类群中只决定花瓣的形成,但在银莲花族中对花瓣和雄蕊的形成都具有一定的功能。所以,小花草玉梅花被片和雄蕊的形态变化都可能与AP3-3基因的改变相关。本研究中花被片自然变异植株除花被片有很大变化外,雄蕊也有一定改变,因此选择AP3-3基因作为该物种变异植株研究的目标基因。本研究利用常规PCR和Hi-Tail PCR方法,从小花草玉梅正常和花被片自然变异植株中分别克隆得到了NArAP3-3和VArAP3-3基因,并运用生物信息学对其序列特征及预测蛋白进行了综合对比分析,以期探索该变异植株中花被片变异产生的可能原因,为进一步探究其变异机制奠定基础。
1材料和方法
1.1材料
2014~2015年6~7月间采集于陕西陇县关山地区的小花草玉梅正常和花被片自然变异植株的花及苞叶,立即冻存于液氮中备用。该花被片自然变异植株命名为绿白相间花变异植株(图1,B)。
菌株E.coliTop10(Tiangen)、克隆载体PMDl9-T(TaKaRa)、PCR Premix(Takara ExTaqTM、CW EsTaq和Tiangen 2×Taq)、DNA提取试剂盒(Tiangen)和胶回收试剂盒(Tiangen)等分别购自相关试剂公司;引物合成在上海生工生物工程公司。
1.2方法
1.2.1小花草玉梅正常植株和绿白相间花变异植株的花部形态学对比以正常和变异植株的花器官标本为材料,将其置于培养皿中的湿润滤纸上,再覆盖1层湿润滤纸,后将培养皿封口,静置3 h后进行花器官各部分分解。分解得到的花被片、雄蕊及雌蕊在尼康体式显微镜下观察,保存结果。
1.2.2小花草玉梅NArAP3-3和VArAP3-3基因中间序列的扩增按照天根DNA提取试剂盒说明书提取小花草玉梅正常植株和绿白相间花变异植株的基因组DNA。然后根据此类基因的保守区域设计引物进行普通PCR扩增,以期获得本材料上此类基因的保守序列。普通PCR扩增引物为F(5′-AT-GGGTAGAGGAAAGATTGAGA-3′)和R(5′-CC-ATTTGACATAGCCATTATAGAT-3′)。反应体系为:预混酶10 μL,F、R引物各1 μL,模板DNA 1 μL,加灭菌去离子水至总体积20 μL。扩增条件为:94 ℃预变性5 min;94 ℃变性30 s,54.5 ℃退火30 s,72 ℃延伸90 s,35个循环;72 ℃延伸10 min。
PCR产物以1%琼脂糖凝胶电泳回收后连接到pMD19-T载体上,42 ℃热激转化大肠杆菌TOP10感受态细胞,经Amp抗性筛选和X-gal/IPTG蓝白斑筛选后选取白色菌落进行液体培养。PCR反应检测阳性克隆,送上海英骏生物技术公司进行测序。
1.2.3NArAP3-3和VArAP3-3基因全长序列的获得根据上面扩增产物得到的测序结果(保守序列)设计巢式引物进行Hi-Tail PCR反应。Hi-Tail PCR扩增引物分简并引物[21]和特异性引物(表1)。本研究直接使用简并引物LAD1-1~LAD1-4与设计的特异性引物进行组合扩增,筛选出在小花草玉梅扩增效果较为理想的随机引物。分别以小花草玉梅正常植株和绿白相间花变异植株的基因组DNA为模板进行Hi-Tail PCR扩增,反应体系和反应条件参考文献[21],1%琼脂糖凝胶电泳检测扩增结果。
将Hi-Tail PCR的第三轮产物进行胶回收,经连接、转化、筛选和鉴定后送公司测序。测序结果利用Primer Premier 5.0、Megalign等软件进行分析,在已知序列的5′端方向上选取3′端与已知片段具有完全相同重叠区域的候选片段进行拼接,而在已知序列的3′端方向上选取5′端与已知片段具有完全相同重叠区域的候选片段进行拼接,并根据所获得的核苷酸序列继续向前步移,直到获得完整的基因序列为止。本实验经3次步移获得了已知序列的5′端侧翼序列,经2次步移获得了已知序列的3′端侧翼序列(表1),从而得到了基因全长。
小花草玉梅正常植株NArAP3-3基因和绿白相间花变异植株VArAP3-3基因克隆时所用引物基本相同,仅在获取5′端侧翼序列的第三次步移中有所不同,前者使用3 R∶SP3-1,而后者使用3 R∶SP3-2;二者进行基因克隆时所用程序也基本相同。
1.2.4NArAP3-3和VArAP3-3基因序列的生物信息学分析Blast搜索正常和花变异植株AP3-3类基因全长序列的同源基因,与本研究所得序列进行多重比对,确定所得基因的CDS序列,运用Gene Structure Display Server 2.0对所得基因全长进行基因结构分析。利用在线网站Http://www.expasy.ch/tools/中的Translate程序将所得CDS序列翻译成氨基酸序列,Blast搜索其同源氨基酸序列,用DNAMAN 8、Megalign软件进行氨基酸序列的多重比对。使用MEGA 5.1软件为该序列与其同源基因在DNA和氨基酸水平上构建邻位相连法的系统发育树,进行Bootstrap检测。选择EXPASY网站及程序进行以下分析:用ProtParam分析翻译蛋白的基本性质;ProtScale对翻译的蛋白进行疏水性分析;SignalIP对翻译的蛋白序列进行信号肽预测分析;TMHMM对翻译的蛋白序列进行跨膜区分析;NetPhos对推导的氨基酸序列进行磷酸化位点预测;应用HNN对所推导的氨基酸序列进行二级结构预测;Phyre2 Server对翻译蛋白序列进行三级结构的初步预测。此外,对本研究得到的2个基因全长进行了启动子分析,所用的分析网站为http://bioinformatics.psb.ugent.be/webtools/plantcare/html/。

表1 Hi-Tail PCR扩增所用引物
Note:V(g,A,C),B(g,T,C),D(g,A,T),N(A,T,g,C).
2结果与分析
2.1小花草玉梅正常植株和绿白相间花变异植株的花部形态学对比分析
正常和变异植株的花器官分解结果(图1)表明,正常植株(图1,A)的花被片共5枚,一轮,形状近椭圆,边缘光滑(图1,G);雄蕊、雌蕊均正常(图1,C、E)。绿白相间花变异植株(图1,B)的花被片共31枚,轮数多轮,外部(图1,H)的5枚花被片颜色完全变绿,中间(图1,I、J)的12枚花被片部分变绿,内部(图1,K、L)的14枚花被片保持白色,这些花被片的形态发生变化且由外向里逐渐接近雄蕊;雄蕊数目减少,部分退化,图1,D中第二排雄蕊均改变,方框所示的3枚雄蕊花药发育不全,其余4枚花丝扁平;雌蕊(图1,F)无明显改变。
2.2小花草玉梅NArAP3-3和VArAP3-3基因的克隆
据保守区域设计引物进行普通PCR扩增,获得部分片段(图2,A、G),测序分析表明该片段为本材料上AP3-3类基因的部分序列。据所得序列设计Hi-Tail PCR反应特异性引物(表1)进行扩增,获得了已知序列5′和3′端的侧翼序列,不断向前步移并相互拼接得到基因全长,获得的基因全长经多次分段单引物验证及不同引物系统重复验证为正确。
在获得基因全长的整个过程中每一次步移都要进行随机引物的筛选,筛选结果及每一次步移的Tail-PCR电泳分析(图2)结果可知:B、C为获得正常植株NArAP3-3基因3′端侧翼序列的2次步移,筛选得到的最佳随机引物分别为LAD1-4和LAD1-3,D、E、F为获得NArAP3-3基因5′端侧翼序列的3次步移,筛选得到的最佳随机引物分别为LAD1-4、LAD1-2和LAD1-2;获得绿白相间花变异植株VArAP3-3基因全长的H、I和J、K、L同理。

A.正常花;B.变异花;C.正常花的雄蕊;D.变异花的雄蕊;E.正常花的雌蕊;F.变异花的雌蕊;
Tail-PCR扩增包括3轮超级PCR反应,B中2和3分别为第一、二轮PCR的产物,4、5、6分别为不同的第三轮引物下该轮PCR的产物;D中10和11分别为第一、二轮PCR的产物,12和13分别为不同的第三轮引物下该轮PCR的产物;K中30为第二轮PCR的产物,31为第三轮PCR的产物;其余的C、E、F、H、I、J和L中的第1孔均为第二轮超级PCR反应的产物,而第2、3孔均为第三轮超级PCR反应的产物且第2、3孔的引物均不同;图中所示的第三轮产物中除箭头标注条带最终成为拼接基因全长序列所用的片段外,其他条带的测序结果均可验证其相应箭头所标注目的带序列的正确性,以确保能得到较为准确的实验结果。
获得基因全长的Blast比对结果表明,两基因分别与不同植物的AP3-3基因有较高一致性,因此分别命名为NArAP3-3基因(正常植株)和VArAP3-3基因(绿白相间花变异植株)。
2.3NArAP3-3和VArAP3-3基因的结构特征
NArAP3-3基因全长3 795 bp,含有长度为1 188 bp(1 344~2 531)的开放阅读框,CDS编码序列为666 bp,编码221个氨基酸,GenBank登录号为KU230445;VArAP3-3基因全长3 898 bp,含有长度为1 188 bp(1 396~2 583)的开放阅读框,CDS编码序列为666 bp,编码221个氨基酸,GenBank登录号为KU230446。
对所得基因全长进行基因结构分析,结果(图3)表明,二者5′端侧翼序列分别为1 343和1 395 bp,开放阅读框均包含7个外显子和6个内含子,外显子所含碱基数依次为188、67、62、103、42、45和159,且与其他植物的AP3-3基因相比,前6个外显子非常保守,所含碱基数基本不变,仅第7个外显子有较大变动,7号外显子构成C结构域,保守性差。此外,它们在第1个外显子上均含有一段120 bp左右DNA结合位点(图3中方框所示)。
2.4NArAP3-3和VArAP3-3基因系统进化分析
由AP3-3类基因氨基酸序列的多重比对可以看出,NArAP3-3和VArAP3-3具有MADS-box基因家族B类亚家族中AP3基因的典型特征,180 bp左右高度保守的MADS结构域,中度保守的K区(植物转录因子的特征结构),保守性差的I区、C端结构域,以及PI衍生结构域和PaleoAP3结构域(图4)。比对18个AP3-3类同源基因DNA全长和氨基酸序列,结果表明,在DNA水平上,NArAP3-3和VArAP3-3基因与大叶铁线莲、长瓣铁线莲的AP3-3基因相似性最高,均为77%,与其他毛茛科植物AP3-3基因的平均相似性分别为71%和70.5%;在氨基酸水平上,NArAP3-3和VArAP3-3与大叶铁线莲的相似性仍最高,分别为75%和74%,与长瓣铁线莲的相似性均为71%,而与其他毛茛科植物AP3-3蛋白的平均相似性分别为66%和64.8%。据DNA全长和氨基酸多序列比对构建系统进化树,从图5,A可以看出,NArAP3-3和VArAP3-3聚在一起,同时也和大叶铁线莲ChAP3-3、长瓣铁线莲CmAP3-3聚在一起且置信度很高,这与相似性结果一致,也与氨基酸序列比对进化树(图5,B)所反映出的基因进化关系基本一致。因此,NArAP3-3和VArAP3-3蛋白均属于PaleoAP3系,NArAP3-3和VArAP3-3基因均属于AP3亚家族中的AP3-3基因。

A~F.获得NArAP3-3基因全长的PCR扩增结果;G~L.获得VArAP3-3基因全长的PCR

方框所示区域为DNA结合位点
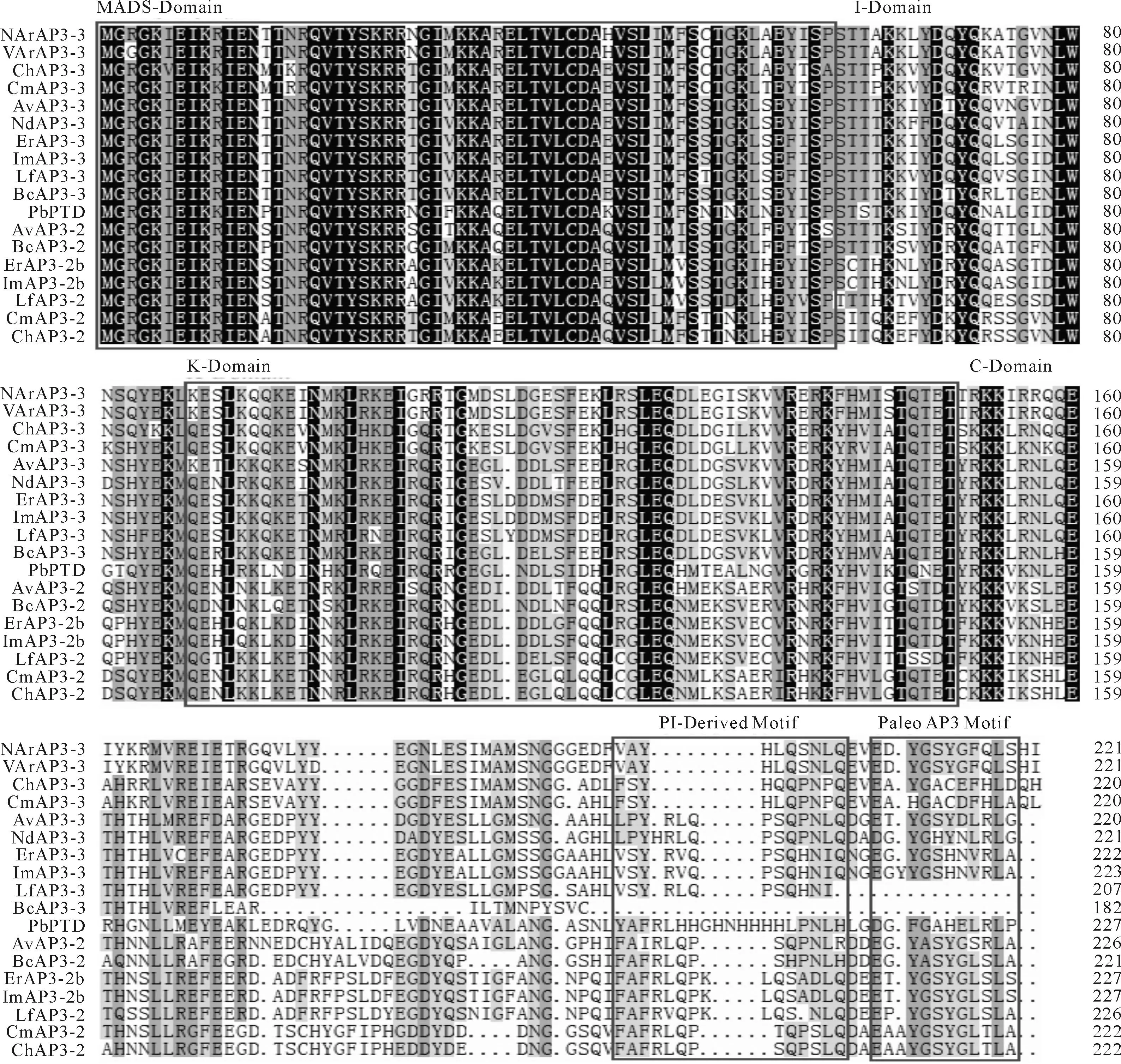
NArAP3-3(KU230445).小花草玉梅正常植株;VArAP3-3(KU230446).小花草玉梅变异植株;ChAP3-3(AGH39918.1)、
2.5NArAP3-3和VArAP3-3基因的启动子分析
对NArAP3-3和VArAP3-3基因的5′端侧翼序列进行启动子分析,可知这2个序列除含有启动子基本元件,如TATA-BOX、CAAT-BOX外,还含有ABA响应元件、真菌诱导应答元件、缺氧诱导型增强子样元件,以及参与生理周期调控、MYBHv1结合位点、干旱诱导中的MYB结合位点和多种光响应元件等一些其他的调控序列,初步推测NArAP3-3和VArAP3-3基因的表达受植物激素、光照、生理周期和MYBHv1转录因子等的调控。
比较这2个基因的启动子分析结果,结合二者的序列对比(图6)可知,NArAP3-3和VArAP3-3基因相比,后者在229~277处有一段49 bp的完全插入序列,与其起始密码子(1 396)相距1 168 bp,由启动子分析结果可知,这段插入序列含有7个CAAT框。CAAT框是存在于启动子及加强区的普通顺式作用原件,显然这49 bp插入序列所涵盖的7个CAAT框位于加强区中,可调控基因表达水平。
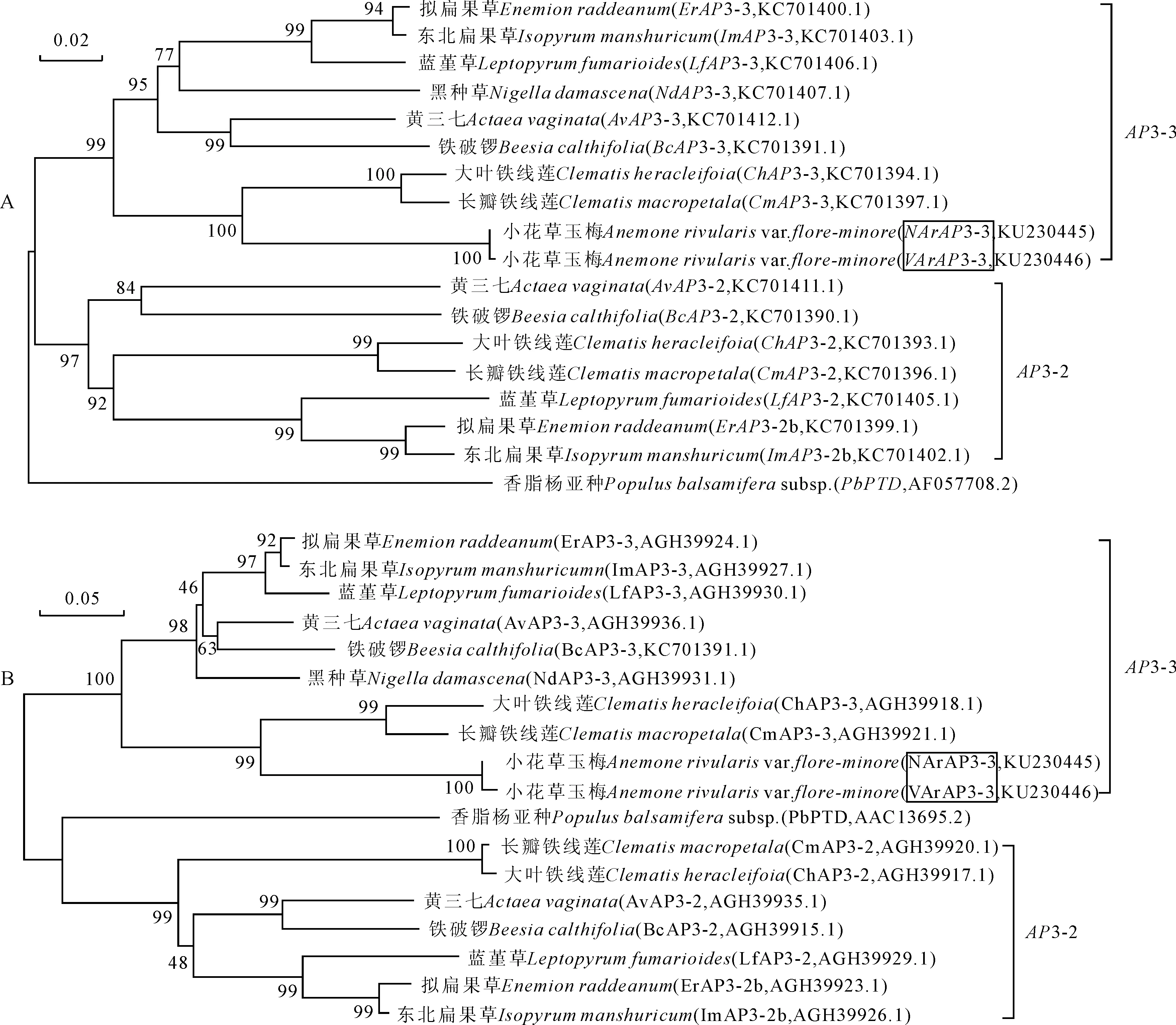
标尺代表遗传距离,各节点处数字表示重复1 000次分析的Bootstrap值;图中加框为本研究克隆所得基因,其他基因名称同图4

A.VArAP3-3与NArAP3-3基因启动子分析结果的差异片段;
2.6NArAP3-3和VArAP3-3基因的氨基酸序列差异及其他相关性质分析
对比二者氨基酸序列知其有2个氨基酸的差异,分别为第3位的R→G(即精氨酸Arg变甘氨酸Gly)和第178位的Y→D(即酪氨酸Tyr变天冬氨酸Asp)。这2个氨基酸的变化直接影响其形成蛋白质的理化性质。
使用各种程序对二者所翻译蛋白质进行蛋白基本性质和疏水性分析及跨膜区、信号肽和磷酸化位点的预测分析。结果表明,二者所翻译蛋白均无信号肽和跨膜区,非分泌蛋白和膜结构蛋白,蛋白质形成后不分泌到胞外而留于细胞内在胞质或核内起作用,这与NArAP3-3和VArAP3-3基因作为MADS-box转录因子基因家族成员,编码转录因子蛋白于细胞核内结合DNA来发挥功能相一致。二者所翻译蛋白的磷酸化位点无显著差别,均为7个Ser位点,8个Thr位点和3个Tyr位点。然而,二者所翻译蛋白的基本性质和疏水性有一定差别:NArAP3-3蛋白分子式为C1121H1824N328O345S11,分子量25.8 KD,预测等电点为9.48(弱碱性蛋白),负电荷残基总数29个,正电荷残基总数39个,不稳定指数57.43(可认为该蛋白不稳定);而VArAP3-3蛋白分子式为C1112H1811N325O346S11,分子量25.6 KD,预测等电点为9.34(弱碱性蛋白),负电荷残基总数30个,正电荷残基总数38个,不稳定指数为59.79(可认为该蛋白不稳定);两蛋白的疏水性分析显示,NArAP3-3蛋白的总平均疏水指数为-0.804,而VArAP3-3蛋白的总平均疏水指数为-0.795,以0为界,正值表示疏水性,负值表示亲水性,疏水指数越大,蛋白疏水性就越强,显然二者均为亲水性蛋白且前者亲水性大于后者。
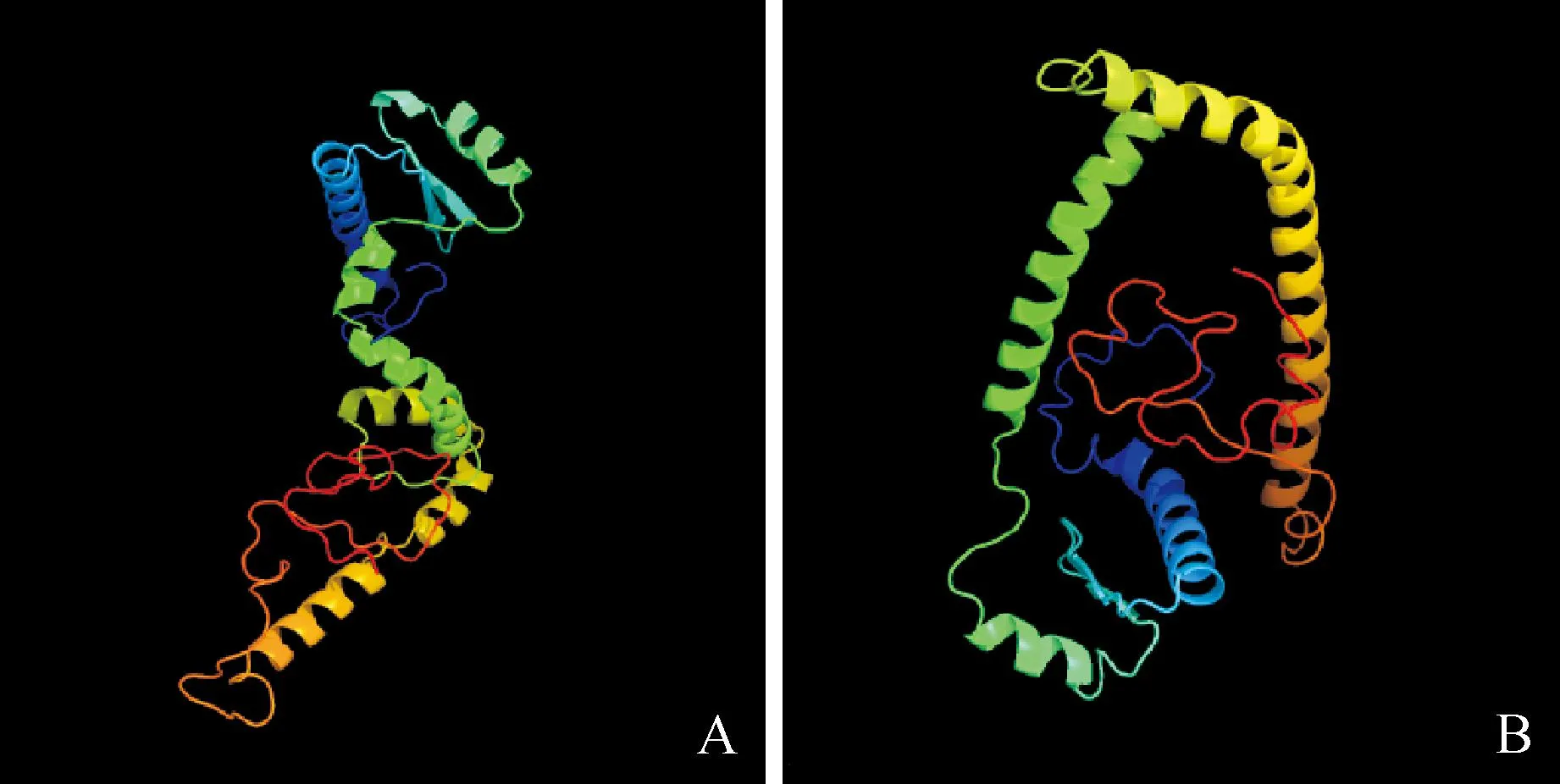
图7 NArAP3-3(A)和VArAP3-3(B)蛋白三维结构预测
2.7NArAP3-3和VArAP3-3蛋白的二级结构及三级结构预测
对NArAP3-3和VArAP3-3基因所推导的氨基酸序列进行二级结构预测,结果有一定差异,即二者的二级结构虽然都只有α螺旋(alpha helix,Hh)、延伸链(extended strand,Ee)和随机卷曲(random coil,Cc)3种结构,但含量略有不同,前者3种结构成分的含量分别为54.30%、13.57%和32.13%,而后者则分别为54.75%、13.57%和31.67%。对NArAP3-3和VArAP3-3蛋白序列进行三级结构预测,其初步的同源建模结果(图7)表明,二者所译蛋白的空间结构明显不同,所以发挥功能的程度有一定差别。
3讨论
经观察,绿白相间花变异植株与正常植株相比,花被片数目、轮数显著增多,形态由外向里逐渐接近雄蕊,且颜色由白逐渐转绿;雄蕊退化,数目减少;雌蕊基本不变。 本研究用常规PCR和Hi-Tail PCR方法克隆得到了小花草玉梅正常和变异植株的AP3-3类基因,分别命名为NArAP3-3和VArAP3-3。DNA全长和氨基酸序列的多重比对及系统发育树分析表明,NArAP3-3和VArAP3-3均与不同植物的AP3-3基因有较高的相似性,都含有典型的MIKC结构且在C末端有PI基序和PaleoAP3基序,故判断NArAP3-3和VArAP3-3为MADS-box家族AP3-3基因的同源基因,与花器官发育相关[14-15]。
VArAP3-3与NArAP3-3基因的序列变化:
(1)VArAP3-3基因在5′上游调控区(229~277处)有一段49 bp的完全插入序列,包含7个CAAT框,位于启动子加强区。位于启动子加强区的插入序列可能直接影响该基因的转录起始频率和强度,调控基因的表达量和表达程度,从而使花器官的表型发生变化。有研究表明,银莲花族铁线莲属内的花瓣缺失机制可能是由调控区变异引起的[22],耧斗菜族扁果草属的花瓣缺失现象也是由EnraAP3-3基因调控序列的缺失引起的[17],说明毛茛科植物的花瓣缺失现象与AP3-3基因的调控区有很大关系。所以小花草玉梅变异植株的花器官变化也可能与VArAP3-3基因的调控区序列改变相关,本研究结果为此提供了一定证据。
另外,研究表明,在植物致病真菌中,目前所发现的靶标基因上调均与启动子区的插入突变有关。这些插入序列中仅有樱桃叶斑病菌和灰霉菌中的插入序列被鉴定为反转座子,而其他植物病原真菌如桃褐腐病菌(65 bp插入),苹果黑星病菌(553 bp插入)和柑橘绿霉病菌(5个126 bp的串联重复和1个199 bp插入)中插入序列的功能属性并无相关信息,调控机制未知[23]。之后发现,某些柑橘绿霉病菌菌株中PdCYP51B基因启动子区199 bp的序列插入,本身能够作为一个强启动子来发挥功能,引起PdCYP51B基因的超表迖而赋予菌株抗药性[23];且CYP51基因启动子区发生的5个126 bp串联重复插入突变,同样可使CYP51基因过表达而产生抗药性[24]。此外,在苹果中,Espley等发现,调控花青素的转录因子基因MYB10上游区域有5个23 bp的正向重复插入,该插入序列能增强MYB10基因的表达,使花青素增加而产生果肉为红色的红苹果[25]。所以,同样地,小花草玉梅自然变异植株的表型变化也可能是由于VArAP3-3基因启动子区的序列插入导致基因表达上调所致。
(2)VArAP3-3的CDS序列与NArAP3-3相比有4个碱基突变位点。VArAP3-3基因CDS序列的碱基突变,造成其推导氨基酸及所译蛋白结构性质的变化,可能在花器官中引起相应的表型变化。VArAP3-3基因推导氨基酸序列的第3位和第178位发生了改变,第3个氨基酸位于MADS结构域中,该区域高度保守,具有与DNA结合、蛋白质二聚体化及与其他蛋白质因子相结合的功能[26],其变化对所译蛋白质的功能有较大影响。本研究结果表明:NArAP3-3和VArAP3-3蛋白无信号肽和跨膜区,既不分泌到胞外也不成为膜结构成分,而作为转录因子蛋白于细胞核内结合DNA来发挥功能;NArAP3-3和VArAP3-3蛋白的分子量、预测等电点、所含正负电荷残基数目都不同,直接影响二者在胞质和核内的运动及蛋白质的稳定性;不稳定指数显示二者所译蛋白都为不稳定蛋白,且花变异植株中VArAP3-3蛋白更加不稳定;总平均疏水指数显示NArAP3-3和VArAP3-3蛋白均为亲水性蛋白且花变异植株中VArAP3-3蛋白的亲水性较正常株中低,说明二者的亲水、疏水性氨基酸含量和分布都不同,直接影响其蛋白质的空间结构,二、三级结构预测结果的差异性可证明这一点。显然,正常植株NArAP3-3和变异植株VArAP3-3蛋白的理化性质、稳定性与蛋白质结构明显不同,这可能影响蛋白活性及其在细胞中的运动、与其他蛋白质因子的结合、对DNA结合位点的识别及与DNA结合的亲和性,进而影响蛋白质功能的发挥而造成花的表型变化。
综上所述,小花草玉梅变异植株中花被片及雄蕊的变化可能与VArAP3-3基因的改变相关。AP3-3是控制花瓣形成的关键基因,小花草玉梅在进化过程中形成的极似花瓣的花被片可能也受花瓣特性基因AP3-3的影响,且Kramer研究[19-20]表明,该基因在银莲花族中影响花瓣和雄蕊的形成,这说明小花草玉梅的变异植株中,花被片及雄蕊的变化可能均与VArAP3-3基因有一定相关性。VArAP3-3基因调控区和编码序列的变化,可能造成其表达量上调及所翻译蛋白的结构性质发生改变,使雄蕊退化减少,退化的雄蕊可能转变成花被片使花被片轮数及数目增多。银莲花族植物的花瓣形态与雄蕊相似而与其他毛茛科植物的花瓣有所区别[22],且本研究中,变异植株增多的部分花被片形态极似雄蕊,均说明雄蕊变为花被片是有可能的;此外,有文献表明,花被片的起源可能晚于雄蕊和心皮,花瓣可能来源于萼片,也可能来源于退化雄蕊[8,14,27],进一步说明雄蕊转变成花被片是有较大可能的。这种初步形成的猜想还需进一步的研究和验证,小花草玉梅材料新颖,变化多样,拥有巨大的探索空间,值得继续探索和挖掘。
参考文献:
[2]GRANDI V,GREGIS V,KATER M M.Uncovering genetic and molecular interactions among floral meristem identity genes inArabidopsisthaliana[J].ThePlantJournal,2012,69(5):881-893.
[3]LIU Z,MARA C.Regulatory mechanisms for floral homeotic gene expression[J].SeminarsinCell&DevelopmentalBiology,2010,21(1):80-86.
[4]COEN E S,MEYEROWITZ E M.The war of the whorls:genetic interactions controlling flower development[J].Nature,1991,353:31-37.
[5]COLOMBO L,FRANKEN J,KOETJE E,etal.The petunia MADS box geneFBP11 determines ovule identity[J].ThePlantCell,1995,7(11):1 859-1 868.
[6]ROUNSLEY S D,DITTA G S,YANOFSKY M F.Diverse roles for MADS box genes inArabidopsisdevelopment[J].ThePlantCell,1995,7(8):1 259-1 269.
[7]HONMA T,GOTO K.Complexes of MADS-box proteins are sufficient to convert leaves into floral organs[J].Nature,2001,409(6 819):525-529.
[8]PELAZ S,GUSTAFSON-BROWN C,KOHALMI S E,etal.APETALA1 andSEPALLATA3 interact to promote flower development[J].ThePlantJournal,2001,26(4):385-394.
[9]THEIβEN G,SAEDLER H.Plant biology:floral quartets[J].Nature,2001,409(6 819):469-471.
[10]KAUFMANN K,MELZER R,THEIβEN G.MIKC-type MADS-domain proteins:structural modularity,protein interactions and network evolution in land plants[J].Gene,2005,347(2):183-198.
[11]ZAHN L M,KONG H,LEEBENS-MACK J H,etal.The evolution of theSEPALLATAsubfamily of MADS-box genes:A preangiosperm origin with multiple duplications throughout angiosperm history[J].Genetics,2005,169(4):2 209-2 223.
[12]DREA S,HILEMAN L C,DE MARTINO G,etal.Functional analyses of genetic pathways controlling petal specification in poppy[J].Development,2007,134(23):4 157-4 166.
[13]KRAMER E M,IRISH V F.Evolution of genetic mechanisms controlling petal development[J].Nature,1999,399(6732):144-148.
[14]THEIβEN G.Development of floral organ identity:stories from the MADS house[J].CurrentOpinioninPlantBiology,2001,4(1):75-85.
[15]DREWS G N,BOWMAN J L,MEYEROWITZ E M.Negative regulation of theArabidopsishomeotic geneAGAMOUSby the APETALA2 product[J].Cell,1991,65(6):991-1 002.
[16]LITT A,KRAMER E M.The ABC model and the diversification of floral organ identity[J].SeminarsinCell&DevelopmentalBiology,2010,21(1):129-137.
[17]ZHANG R,GUO C,ZHANG W,etal.Disruption of the petal identity geneAPETALA3-3 is highly correlated with loss of petals within the buttercup family (Ranunculaceae)[J].ProceedingsoftheNationalAcademyofScienceoftheUnitedStatesofAmerica,2013,110(13):5 074-5 079.
[18]常鸿莉,任毅,冯鲁田.小花草玉梅变态花萼片的形态学研究[J].植物分类学报,2005,43(3):225-232.
CHANG H L,REN Y,FENG L T.Morphological observations on metamorphosed sepals inAnemonerivularisvar.flore-minore(Ranunculaceae)[J].ActaPhytotaxonomicaSinica,2005,43(3):225-232.
[19]RASMUSSEN D A,KRAMER E M,ZIMMER E A.One size fits all?Molecular evidence for a commonly inherited petal identity program in Ranunculales[J].AmericanJournalofBotany,2009,96(1):96-109.
[20]KRAMER E M,DI STILIO V S,SCHLÜTER P M.Complex patterns of gene duplication in theAPETALA3 andPISTILLATAlineages of the Ranunculaceae[J].InternationalJournalofPlantSciences,2003,164(1):1-11.
[21]LIU Y G,CHEN Y.High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences[J].BioTechniques,2007,43(5):649-656.
[22]杜青高.升麻族(Cimicifugeae)植物花瓣缺失分子机制的初步研究[D].西安:陕西师范大学,2011.
[23]孙学鹏.柑橘绿霉病菌比较基因组及抗DMI杀菌剂机理研究[D].杭州:浙江大学,2015.
[24]HAMAMOTO H,HASEGAWA K,NAKAUNE R,etal.Tandem repeat of a transcriptional enhancer upstream of the sterol 14alpha-Demethylase gene (CYP51) inPenicilliumdigitatum[J].AppliedandEnvironmentalMicrobiology,2000,66(8):3 421-3 426.
[25]ESPLEY R V,BRENDOLISE C,CHAGNÉ D,etal.Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples[J].ThePlantCell,2009,21(1):168-183.
[26]SHORE P,SHARROCKS A D.The MADS-box family of transcription factors[J].EuropeanJournalofBiochemistry,1995,229(1):1-13.
[27]PELAZ S,DITTA G S,BAUMANN E,etal.B and C floral organ identity functions requireSEPALLATAMADS-box genes[J].Nature,2000,405(6 783):200-203.
(编辑:宋亚珍)
Cloning and Sequence Analysis ofAP3-3 Gene in Normal Plant and Natural Variant fromAnemonerivularisvar.flore-minore
ZHANG Ting,XING Ni,WANG Chao,LIU Huqi*
(College of Life Science,Northwest A&F University,Yangling,Shaanxi 712100,China)
Abstract:The study takes normal plant and tepal natural variant which have big differences in appearance as materials from wild Anemone rivularis var.flore-minore.The class B genes were isolated from genomes of normal plant and natural variant by using conventional and Hi-Tail PCR techniques.They were proved to be AP3-3 branch of paralogs which belong to AP3 gene family from B class MADS-box genes by sequence analysis and named NArAP3-3 (in normal plant) and VArAP3-3 (in natural variant),respectively.The full length of NArAP3-3 gene was 3 795 bp whereas 3 898 bp in VArAP3-3,they both have a 666 bp open reading frame (ORF) and can encode 221 amino acids.The protein showed a typical MADS-box gene structure containing MADS domain,K domain,Ⅰ region and C terminus.The comparison result of full length sequences between NArAP3-3 and VArAP3-3 genes was found to have a 49 bp insertion in VArAP3-3 gene,and there are also 4 base mutations in ORF sequence compared with the NArAP3-3 gene.By bioinformatics analysis of the two’s full length sequences,221 amino acids and insertion sequence,we can find that there are differences in all aspects of gene promoter,basic properties of protein,structure function domain,advanced prediction structure and so on.These differences may be one of the causes of tepals variation,and this study lays the foundation for the further exploration of its variation mechanism.
Key words:Anemone rivularis var.flore-minore;tepals variation;Hi-Tail PCR;MADS-box gene;NArAP3-3 and VArAP3-3 genes
中图分类号:Q754
文献标志码:A
猜你喜欢
中国农学通报(2022年4期)2022-03-02
浙江林业(2021年8期)2021-09-22
花生学报(2019年2期)2019-10-22
小天使·一年级语数英综合(2019年4期)2019-10-06
小学生优秀作文(低年级)(2018年3期)2018-08-15
花卉(2017年17期)2017-10-12
Coco薇(2017年5期)2017-06-05
生物学教学(2017年6期)2017-02-18
琴童(2016年7期)2016-05-14
红蜻蜓·低年级(2016年9期)2016-05-14
