
仿制药质量和疗效一致性评价工作建议*
2016-03-06 02:04葛广波吴敬敬
世界科学技术-中医药现代化 2016年5期
宁 静,葛广波,吴敬敬,杨 凌
(中国科学院大连化学物理研究所 大连 116023)
仿制药质量和疗效一致性评价工作建议*
宁 静,葛广波,吴敬敬,杨 凌**
(中国科学院大连化学物理研究所 大连 116023)
目前,中国医药产业正处于蓬勃发展期,国家在此时推行仿制药质量一致性评价对提升中国制药行业整体水平,保障公众用药安全等具有重大意义。本建议针对当前中国制药行业在药品质量一致性评价中面临的困惑,结合国外药品评价相关制度和经验,从药品分类及其申报原则、药品终点评价指标及其科学内涵、仿制药生物等效性评价相关原则等方面进行了细致剖析,并归纳总结了药品质量一致性和优劣性的评价原则及注意事项。此外,作者还针对中国制药行业的现状和存在的一些具体问题,提出了仿制药质量和疗效一致性评价的一些具体工作建议。
仿制药 新药申报 一致性评价 终点指标 生物等效性
近年来,中国医药产业在研发、生产和监管等方面均有长足发展。特别是近期国家食品药品监督管理局下定决心要解决医药监管领域尤其是仿制药监管上的遗留问题,这使我们对中国医药产业的发展充满信心。此次中国开展仿制药质量一致性评价的核心是提高药品的质量,并通过此次改革促进医药行业产业的结构调整和转型升级,实现上市产品的有效性、安全性和质量可控性,能够达到国际先进水平,满足公众的用药需求。
在医药监管领域,发达国家的先进经验值得认真学习。我们需要大量引进西方发达国家的专利过期药物,从这些药物的研发、生产、评审和监管中,可以充分、合理、有效地借鉴国外的相关经验和原则,全面提升中国医药领域各方面的水平,促使中国逐步由落后、比肩、再到赶超国际先进水平。
从监管科学的角度来讲,我们需要树立一个总体的科学原则,将仿制药的与新药研发的“评价和监管”纳入科学、统一的原则体系中。无论是新药还是仿制药,包括已上市药物和拟上市的药物都应接受横向的相互比较,保障已上市药物在市场中的安全性、有效性和优质性等。从这个层面上来说,出台的原则不应是“运动式”的,更不能在科学上有“原则性”和“逻辑性”的裂痕。
1 药品申报分类原则
一个完善的新药评价体系应该有其统一的科学原则、逻辑和连续性。根据市场上药物“有无”和“优劣”比较原则,可以将药物申报分成全新、改良和仿制3类。申报上市的药物都需要与已上市药物进行横向比较,该过程可促进中国药物研发、制造、评价和监管水平不断提升,保证中国医药行业具有“优胜劣汰”、“良性循环”的新陈代谢能力。
中国现阶段的药物研发水平基本处于改良或者仿制水平,改良药物的研发建立于固定研发模式的基础上。尽管由新化合物(新母体或者新衍生物)产生的治疗药物被视为新药,但仅仅利用新化合物、仍然沿用已有研发“路径和模式”获得的产品,其创新性大打折扣。随着全球药物研发能力的提高,医疗水平的进步,在相同治疗领域,现有上市药物产品已经出现高度重叠的现象。
从科学的角度而言,不以新靶点或者新适应症(市场上“有无”药物)来分类就不能算创新,尽管是新化合物,这样的药物最多能被称之为“改良”药物。坚守这一原则能帮助杜绝“貌似”新药,但实质为仿制或改良的“新药产品”。否则,由于指导原则的“漏洞”,国家医药监管机构将花费大量精力去应对这种仿制“新药”。
如表1所示,把药物的适应症(靶点)、活性成分或者活性部位、给药路径、剂量、规格、非活性部位如辅料共六方面的新颖性作为标准,全新的适应症或者靶点显然应归为全新的“新药”;新的活性成分、活性部位及给药路径归为“改良”药物,因为适应症没发生变化的情形下,比较的显然是药物的“优劣性”;药物在剂量、规格、非活性部位如辅料的变化应该归入“仿制”的类别。
2 药品终点评价原则
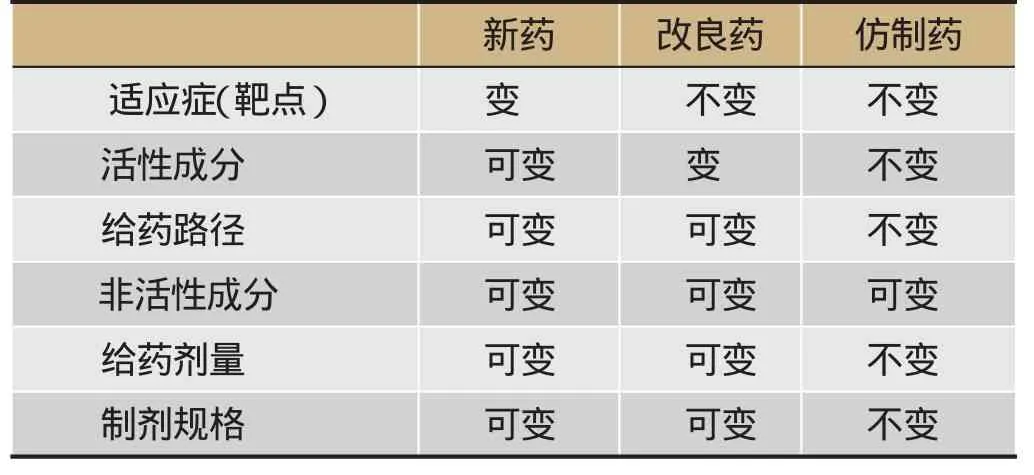
表1 药物的分类原则
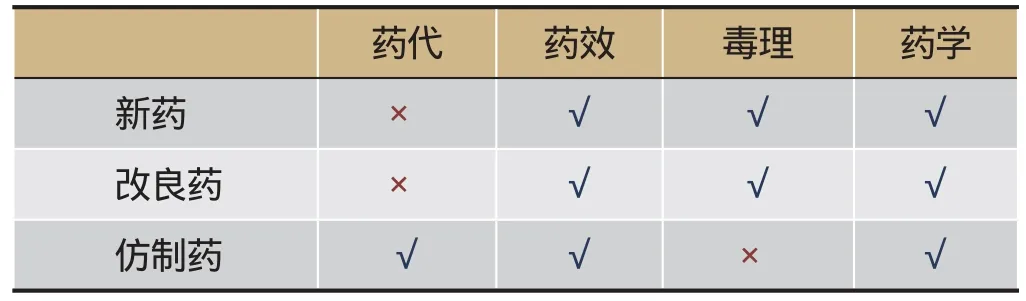
表2 药物终点评价原则
药品的评判有四大“终点评判”层次:药学、药代、药效及毒性。监管机构应该出台经过科学公认的“终点评判”指导原则,有关具体操作或技术过程不应僵化,应该尽量采用建议(而不是必须遵从)性指南或者公开征集专家的意见,专家应以其学术论文和成果作为话语权根据(见表2)。
在改良和仿制药物中,特别是仿制药物,目前的一致性评价最终原则仍然是治疗等效性。然而,药物的体内暴露量(而非药物剂量)才是真正与药效呈现“量效关系”的物质基础,因此只有剂量的“药学评价”不能作为唯一的“终点评判”标准。
早在1984年,FDA就已经出台了以药代动力学属性(生物等效性)作为“终点评判”标准的指导原则[12]。生物等效性研究是一种比较两种相同药物产品的药代动力学终点判断的临床试验研究。美国食品药品管理局(Food and Drug Administration,FDA)和欧洲药品管理局(European Medicines Agency,EMA)是以“待测药物与参比药物之间药代动力学终点的Cmax和AUC平均平方根比值90%的可信区间落入0.8-1.25之间”为标准的。实验设计选择通过两序列的交叉实验设计和足够的Wash-out时间,以保证两种剂型成为“镜像”式比较[3,4]。
与治疗等效性相比,药代动力学终点评判可以节省仿制药大量的研制成本,以量效关系为科学依据,具有“桥接”药学评价与治疗等效性评价的能力[5]。迄今为止,该原则已在美国使用超过30年,许多发达国家也已效仿了20余年,积累了大量的经验。
在新药申报原则中,药学、药效、毒性这3个层次的评价一直是作为“终点评判”标准来使用的[6],但通常药代属性未被设立为新药申报中的终点评判原则。没有将药代属性优劣性作为终点评判直接导致大量的高变异药物(Highly Variable Drugs)得以上市[7,8]。据文献报道,高变异药物占据上市药物的31%,其中,60%的高变异药物源于其药代动力学特性[9]。高变异药物增加药物使用的风险性,使药物副作用和药-药相互作用频发。而药物副作用是导致住院患者致死的第3大原因[10]。面对临床用药风险层出不穷的挑战,通常FDA的补救方式是将药代属性的“不良”以药品说明书标签的方式加以警示,为医生用药增加血药浓度等指导性监测[9]。然而,这些手段都过于被动,时下流行的“精准医学”中的“精准药学”也是不得已的应对手段[11]。显然,这样的方式是用大幅度增加医疗费用和成本来换取或抵抗高风险成本,对于发展中国家来说,这难于为继。
总而言之,如不将药代动力学属性的好坏直接作为终点评判标准是要付出代价的,我们应该从西方的经验中汲取教训,改变对药物药代属性评价研究的轻视。改变对于药物药代属性的基本认识,将其提高到与“药学、药效和安全性”这3个层次的评价等同的认识水平,杜绝中国医药产品中劣质药物的层出。
3 生物等效性评价原则
仿制药的“终点评判”国际上目前通行基于药代动力学性质的优劣作为标准,以原研药为基准,比较仿制药的相对生物利用度(Relative Bioavailability)或生物等效性试验(Bioequivalence)[1,12]。其科学本质是仿制药如果与原研药相似,其在人体内暴露数率和程度(Cmax/Tmax和AUC)就应该一致。
在新药研发中,由于药代属性评判未作为终点评判标准,因此,通过新药申报上市的药物,其药代属性未必“良好”。这给仿制药的评价带来巨大挑战[7]。
此外,该评判是以表观药代属性的一致性为标准的。因此,当受试药物的药学相关指标全部通过时,能否或多大程度保证其与原研药的人体药代属性的一致性?依据BCS分类进行一致性评价的科学内涵?直接进行生物等效性试验其风险有多大?会有哪些挑战和科学问题?表观药代行为与药代属性的决定因素的关系?药代动力学学科的研究水平在此时就显得极其重要。
而仍需要说明的是,目前对仿制药的质量评价通常是建立在基本生物等效性假设的基础之上的,即如果两种药物制剂具有生物等效性,则它们就能够达到质量等效。然而,生物等效性并不是治疗等效的唯一必要条件,而治疗等效的前提也未必是生物等效。因此,在未进行临床治疗等效性评价时,对仿制药的质量评判也常常颇有争议[13]。
通过生物等效性研究评判仿制药一致性可能存在的情况:
(1)药物生物等效,同时治疗等效
(2)药物生物不等效,但治疗等效
(3)药物生物等效,但治疗不等效
(4)药物生物不等效,治疗亦不等效
考虑到“(2)”,则应该给予仿制药研发公司申述及追加治疗等效性评价的机会。而“(3)”则可能成为原研药研发公司反驳监管机构准许仿制药上市的说辞,在该情况下,则可能需要完成治疗等效性评价实现最为权威的评价。
在仿制药一致性评价工作中,药物生物等效性和治疗等效性之间的矛盾是普遍存在的,可能导致一系列法律问题。而中国尚未有相关条例对此给予明确的说明和规范,亟需构建一套完善的法案进而规避可能引起的争端。
4 具体建议
(1)扩充专家组成员,引入药代动力学专家;
(2)征集药代属性及ADME属性研究和评价的方法,特别是体外人源性ADME属性研究和风险评价方法;
(3) 建立临床I期规范性及方法,完善相关指导原则;
(4)征集实施“治疗等效性”合理方案的建议;
(5)建立合理的申述机制,如生物等效性研究不合格或失败时,厂家仍可申请补充追加进行治疗等效性研究的权利等。
1 Drug Price Competition and Patent Term Restoration Act of 1984. United States Federal Law. Public Law,98-417.
2 Sokal M,Gerstenblith B A. The Hatch-Waxman Act: encouraging innovation and generic drug competition. Curr Top Med Chem,2010,10(18): 1950-1959.
3 EMA,Guideline on the Investigation of Bioequivalence. Committee for Medicinal Products for human Use,2010.
4 FDA. Guidance for Industry,Bioavailability and Bioequivalence Studies for Orally Administered Drug Products-General Consideration. Center for Drug Evaluation and Research,2003.
5 LionbergerRA. FDA Critical Path Initiatives: Opportunities for Generic Drug Development,AAPS J. 2008,10: 103-109.
6 And LL,Atkinson J Aj. Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria,validation,strategies.Lesko and Atkinson,Ann Rev PharmacolToxicol,2001,41(41): 347-366.
7 Blume H H,Midha K K. Bio-International 2,Bioavailability,Bioequivalence and Pharmacokinetic Studies. J Pharm Sci,1993,82(11): 1186-1189.
8 Shah V P,Yacobi A,Barr W H ,et al. Evaluation of orally administered highly variable drugs and drug formulations. Pharm Res,1996,13(11): 1590-1594.
9 Davit B M,Conner D P,Fabian-Fritsch B,et al. Highly Variable Drugs: Observations from Bioequivalence Data Submitted to the FDA for New Generic Drug Applications. AAPS J,2008,10(1): 148-156.
10 Lazarou J,Pomeranz B H,Corey P N. Methods of using zonisamide as an adjunctive therapy for partial seizures. JAMA,1998,279(15): 1200-1205.
11 Xu J,Gong B,Wu L,et al. Comprehensive Assessments of RNA-seq by the SEQC Consortium: FDA-Led Efforts Advance Precision Medicine. Pharmaceutics,2016,8(1).pii: E.
12 Meersch AVD,Dechartres A,Ravaud P. Quality of reporting of bioequivalence trials comparing generic to brand name drugs: a methodological systematic review. PLoS One,2011,6(8): e23611.
13 Chow S C. Bioavailability and Bioequivalence in Drug Development. Wiley Interdiscip Rev Comput Stat,2014,6(4): 304-312.
A Suggestion for Qualityies of Generic Drugs and the Therapeutic Equivalence Assessment
Ning Jing,Ge Guangbo,Wu Jingjing,Yang Ling
(Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,China)
At present,the pharmaceutical industry in China is undergoing the state of vigorous development. It is a critical to improve the equivalence assessment of drug quality,in order to press ahead with the development of the pharmaceutical industry as well as strengthen the medication security and safety. This article focused on the perplexity of the drug quality and therapeutic equivalence assessment,combined with both domestic and foreign evaluation regulation and experience and conducted a profound analysis on the drug classification and relative application regulations,evaluation end points,scientific implications,bioequivalence assessment and relative regulations. In this study,the evaluation regulations and considerations was summarized. At last,aiming at the specific questions of the pharmaceutical industry in China,several suggestions for qualities of generic drugs and their therapeutic equivalence assessments were provided.
Generic drugs,new drug application,equivalence assessment,end point,bioequivalence
10.11842/wst.2016.05.003
R284
A
(责任编辑:朱黎婷,责任译审:朱黎婷)
2015-04-20
修回日期:2016-04-20
* 科学技术部“重大新药创制”科技重大专项(2012ZX09506001006):早期ADME/Tox关键技术研究,负责人:杨凌;国家自然科学基金委面上项目(81573501):细胞色素P450 3A亚型酶与配体间相互作用的结构和机制研究,负责人:杨凌;国家自然科学基金委青年基金项目(81503152):蟾蜍甾烯类化合物结构-代谢稳定性关系研究,负责人:宁静。
** 通讯作者:杨凌,本刊编委,博士,研究员,主要研究方向:药物代谢、药物早期成药性评估及新药研发。
猜你喜欢
化工管理(2022年14期)2022-12-02
昆明医科大学学报(2021年6期)2021-07-31
疯狂英语·新悦读(2020年1期)2020-02-20
自动化学报(2019年6期)2019-07-23
中国医院院长(2017年7期)2017-06-15
文学教育(2016年27期)2016-02-28
转化医学电子杂志(2015年4期)2015-12-27
创业家(2015年9期)2015-02-27
创业家(2015年9期)2015-02-27
中成药(2014年9期)2014-02-28
