
G蛋白偶联受体的结构生物学研究
2016-03-06 08:27:41张浩楠吴蓓丽
自然杂志 2016年3期
张浩楠,吴蓓丽
中国科学院上海药物研究所中科院受体重点实验室,上海 201023
G蛋白偶联受体的结构生物学研究
张浩楠,吴蓓丽†
中国科学院上海药物研究所中科院受体重点实验室,上海 201023
G蛋白偶联受体(G protein-coupled receptor, GPCR)在细胞信号传导过程中发挥关键作用,其三维结构的解析对于深入理解GPCR的结构与功能关系具有重要意义。2000年以前,GPCR的高分辨率结构解析一直是困扰科学家们的一个难题。近年来,GPCR的结构生物学研究实现了飞跃式的发展。本文简要综述了GPCR结构解析的方法与创新点,并以CCR5和P2Y1R两种受体的结构解析为例阐述GPCR结构对于功能研究和药物研发的重要性。
G蛋白偶联受体;晶体结构;CCR5;P2Y1R
生物体内包含一系列复杂而精细的生理信号传导通路,形成庞大的信号传导网络,参与调节各项生理平衡从而保证生物体内环境稳态。其中,GPCR参与了生物体内大部分的生理活动和信号调节,是迄今为止发现的最大的受体超家族,由超过1 000种受体蛋白组成。GPCR具有七次跨膜螺旋结构,其胞外区域结合细胞外信号分子(气味、激素、神经递质等),胞内区域招募并结合下游信号分子[1](G蛋白或β-arrestin效应蛋白、第二信使等)。下游信号分子主要通过cAMP信号通路和磷脂酰肌醇信号通路参与调节体内多项生理活动。GPCR功能失调会导致多种疾病的产生,如心脑血管疾病、代谢疾病、神经相关性疾病、视觉相关疾病、炎症、免疫性疾病以及癌症等,因此GPCR是人体内重要的药物靶标[2]。据统计,目前已上市的药物中有40%以上作用于GPCR,其中著名的药物包括奥氮平、氯雷他定、雷尼替丁、替加色罗等。
根据GPCR氨基酸序列的相似性,可将该受体家族分为四个亚家族,分别是类视紫红质受体(A家族)、分泌素受体和粘附素受体(B家族)、代谢性谷氨酸受体(C家族)和味觉受体(F/TAS家族)[3]。其中A家族包含的受体数量最多,超过700个,调节人体内大部分的生理活动,是人体内参与调节信号通路最多的GPCR亚家族。
1 GPCR结构解析中的方法与创新
由于蛋白表达量低、稳定性差、多种构象共存等原因,GPCR的结构解析一直是极具挑战性的科学难题。直到2000年,第一个哺乳动物体内GPCR——牛视紫红质蛋白的结构被成功解析,人类第一次在原子水平看到了GPCR的三维结构[4]。尽管如此,成功解析一个GPCR结构仍需要攻克在蛋白表达、纯化和结晶过程中的许多难题。随着近年来膜蛋白研究技术的发展,GPCR的结构研究得到了突破性的进展[5]。下文将简单阐述GPCR结构解析中的主要方法与创新点。
1.1 GPCR蛋白表达
GPCR在生物体内表达量低,从天然组织中难以获得足够量的GPCR蛋白用于结构解析。为了得到足够量的GPCR蛋白样品进行蛋白结晶实验,必须使用重组蛋白表达系统进行蛋白表达。常用于蛋白表达的系统主要包括大肠杆菌(E. coli)、酵母、昆虫细胞和哺乳动物细胞[6]。大部分结构已解析的GPCR使用昆虫细胞表达系统进行蛋白表达。该表达系统与大肠杆菌表达系统相比,蛋白翻译后修饰比较接近哺乳动物细胞水平,可以更好地模拟人体细胞内的天然环境,并且可以高量表达目的蛋白[7]。虽然哺乳动物细胞更接近人源细胞,但哺乳动物细胞表达蛋白的糖基化修饰复杂,增加了后续实验中去除糖基的难度,而去除糖基化不充分将会阻碍GPCR蛋白分子在脂立方相中的规则排列,影响其堆积形成晶体。以上问题成为哺乳动物细胞表达系统用于GPCR结构解析的限制因素。在酵母细胞表达系统中,蛋白的N-糖基化修饰不同于昆虫细胞[8],而以往研究表明正确的N-糖基化对于GPCR的正常表达及运输上膜非常必要[9-10]。因此,昆虫细胞是目前最常用于GPCR蛋白表达的表达系统。
1.2 GPCR蛋白稳定性的提高
GPCR作为膜蛋白,其大部分分子表面疏水性强,在水溶液中易聚沉[11],因此采用去污剂包裹GPCR蛋白分子的疏水部分形成微团以防止其聚集[12]。即使在去污剂包裹的微团里,GPCR分子的构象依然很不稳定,可处于多种构象状态,阻碍了晶体形成。获得高稳定性的GPCR蛋白样品是解析其结构的先决条件。
提高GPCR蛋白稳定性的常用方法包括:第一、利用高亲和力的特异性配体,将GPCR蛋白分子稳定在某个单一构象状态,使其更易于规则排列堆积形成蛋白晶体[13-14]。第二、在GPCR构象比较灵活的胞内环区域或胞外N端区域插入可溶蛋白片段帮助稳定GPCR构象[14-15],并提高GPCR蛋白产量。同时,融合蛋白可增大蛋白的亲水表面积,辅助GPCR蛋白分子之间的相互作用,促进晶体堆积。目前比较成功的融合蛋白包括T4溶菌酶蛋白、B256RIL[16]等。近年来越来越多适宜稳定GPCR构象的融合蛋白被发掘。第三、在GPCR分子中引入某些氨基酸突变,可增强GPCR分子内部的相互作用力(包括氢键、盐桥、二硫键、疏水作用等),从而提高蛋白稳定性[17-18]。另外,目前已有研究表明抗体和纳米抗体同样可以稳定GPCR蛋白分子辅助其结晶[19]。利用上述方法获得高稳定性的GPCR蛋白样品,即可进行蛋白结晶实验。
1.3 GPCR蛋白结晶
X射线晶体衍射技术是目前用于GPCR结构解析的主要方法。利用传统的气液扩散结晶方法很难获得高质量的GPCR蛋白晶体,目前主要利用脂立方相膜蛋白结晶技术进行GPCR蛋白结晶实验,该方法由Landau等人于1966年提出[20]。脂立方相是将脂和水溶液按一定比例混合形成的在空间上连续的水通道和脂质相,GPCR蛋白分子在脂质相中移动进行晶体堆积。脂立方相模仿了天然的细胞膜脂双层环境,可显著提高GPCR蛋白的稳定性,并降低了结晶实验对蛋白量的需求。
2 GPCR的晶体结构解析
近年来,GPCR晶体结构的研究取得了一系列突破性的进展。2007年,第一个人源GPCR——β2肾上腺素受体的晶体结构被解析,实现了人源GPCR结构解析零的突破[14,21]。仅在2012年一年内就有9种GPCR的晶体结构被解析,至今已有超过30种GPCR的晶体结构被解析,极大地促进了人们对于GPCR三维结构的认识(表1)。这些结构已解析的GPCR在结构和功能上具有丰富的多样性,既包括A家族的肽类受体、脂类受体等,也有B家族的胰高血糖素受体,C家族的SMO受体。所解析的大部分是GPCR与拮抗剂或反向激动剂结合的复合物结构,展示了处于非激活状态的受体构象。Rhodopsin、β2AR、A2AAR、P2Y12R等GPCR分别与拮抗剂和激动剂结合的复合物结构已被解析,显示了GPCR蛋白从非活性状态向活性状态转变的受体构象变化。不同GPCR的配体在大小、形状和电荷性质上存在着很大的差异,已解析的GPCR结构显示了不同GPCR蛋白与各自配体的特异性结合模式,为不同GPCR识别性状迥异的配体分子提供了结构基础。从配体进入途径来说,大部分GPCR的配体都是通过胞外区直接进入开放的配体结合口袋与GPCR相互作用,而脂类受体的配体结合口袋则被蛋白N端区段或胞外环区封闭,其配体很可能经由脂分子双层进入配体结合位点。

表1 结构已解析的GPCR
2011年,β2AR和G蛋白异源三聚体的复合物结构被解析,人们第一次看到了处于激活状态的GPCR蛋白是如何与重要的下游信号蛋白G蛋白相互作用的[22]。2015年视紫红质(Rhodopsin)和β抑制蛋白(β-arrestin)复合物结构的解析,则将GPCR与另一条信号通路中效应蛋白的结合模式呈现在人们的面前[23]。这些GPCR结构的解析使人们更加深入地了解GPCR在胞内的信号传导机制。
GPCR作为人体内调节多种生理活动的重要膜蛋白家族,仍有许多复杂的结构特点需要进一步研究,例如偏向性配体对受体活性的调节机制、变构调节剂对受体构象的影响、GPCR信号通路下游其他信号蛋白的招募以及非A家族GPCR的信号识别机制等,这些问题都有待于继续深入研究。因此,对GPCR的进一步结构研究仍具有十分重要的意义。
我们解析的趋化因子受体CCR5和嘌呤能受体P2Y1R的晶体结构阐明了这两种GPCR受体与特异性配体分子的精细作用机制,为艾滋病和血栓性疾病的新药研发提供了结构基础和线索。下面,我们将简要介绍CCR5和P2Y1R的晶体结构。
2.1 趋化因子受体CCR5的晶体结构
趋化因子受体CCR5在巨噬细胞、树突状细胞及记忆T细胞等多种免疫细胞中表达,在免疫应答调控中结合巨噬细胞炎症因子1α(MIP-1α)[24]、巨噬细胞炎症因子1β(MIP-1β)及单核细胞趋化蛋白2(MCP-2)等多种趋化因子[25-26],激活并调控T细胞和单核细胞、巨嗜细胞系的迁移、增殖,因此CCR5在免疫应答中发挥着重要作用[27]。此外,CCR5与艾滋病的发生息息相关。CCR5和另一种趋化因子受体CXCR4可作为HIV病毒的共受体,辅助病毒侵染宿主细胞[28]。不同类型的HIV病毒选择性结合不同共受体以感染人体细胞。根据对共受体的选择性,HIV病毒被分成三种类型,分别是R5嗜性、X4嗜性以及R5X4嗜性[29]。2010年,CXCR4分别与两种拮抗剂结合的复合物晶体结构被解析[30]。2013年,我们解析了CCR5与抗HIV病毒药物马拉维若结合的复合物晶体结构[31]。CXCR4和CCR5的晶体结构对于深入研究HIV病毒的感染机制及其他生物学功能提供了重要的结构基础。
HIV病毒荚膜糖蛋白gp120的第三可变环区域(V3 loop)是结合共受体CXCR4和CCR5的关键位点。将不同HIV病毒gp120的V3 loop进行氨基酸序列比对,发现X4嗜性病毒的V3 loop含有较多的正电性氨基酸[32],而CXCR4的晶体结构显示其配体结合口袋内含有较多的负电性氨基酸[30](图1(b)),例如受体跨膜螺旋以及胞外环上的天冬氨酸,而这些氨基酸也是影响HIV病毒入侵的关键氨基酸[30]。与CXCR4不同,CCR5配体结合口袋内的氨基酸带电荷较少(图1(c))。这些不同电性氨基酸在受体CXCR4和CCR5的分布分别与X4嗜性病毒、R5嗜性病毒V3 loop的氨基酸的电荷分布相关,说明了共受体配体结合口袋内的电荷分布对于选择性识别不同种类的HIV病毒具有重要作用。此外,基于CCR5和CXCR4分别与R5嗜性病毒和X4嗜性病毒gp120的分子对接模型,我们进一步发现不同共受体的配体结合口袋内关键氨基酸的侧链长短不同造成了空间位阻的巨大差异,是导致CXCR4和CCR5选择性识别不同HIV病毒的另一重要因素[33-34]。这些发现为我们深入理解HIV病毒与共受体的特异性结合模式提供了结构依据。
马拉维若是目前美国食品药物监管局(FDA)批准上市的唯一以CCR5为靶点的抗HIV病毒感染药物[35],以往的研究表明马拉维若是CCR5的变构调节剂[36-37],但其作用机制一直未能被明确阐明。数据表明CCR5的天然配体——HIV病毒糖蛋白gp120和趋化因子等主要与CCR5受体的N端区域和第二胞外环结合[38-39],而CCR5结构显示马拉维若的结合位点较深,与CCR5天然配体的结合区域不同,其主要位于由跨膜螺旋构成的结合口袋内(图1(a)),因此马拉维若无法像正构配体那样通过形成空间位阻阻断CCR5与其天然配体的结合。我们在CCR5与马拉维若的复合物结构中发现,两个在A家族GPCR受体中高度保守的氨基酸Trp248和Tyr244的构象与其他已解析的GPCR非活性构象相似并区别于处于活性状态的GPCR结构,而已有研究表明这两个保守氨基酸在GPCR受体激活时会发生构象改变[22,40],这表明CCR5与马拉维若结合时其构象处于非活性状态。此外,马拉维若分子中的苯环与受体的氨基酸Trp248形成疏水作用,进一步稳定了CCR5的非活性构象。综上所述,马拉维若能够阻断HIV病毒结合CCR5,并非依赖于马拉维若的空间位阻作用,而是将CCR5的构象稳定在非活性状态,降低了CCR5与HIV病毒糖蛋白gp120的结合能力,从而抑制了病毒感染。CCR5结构揭示了药物分子马拉维若的抗病毒机制,对基于结构的抗HIV药物研发具有重要的意义。
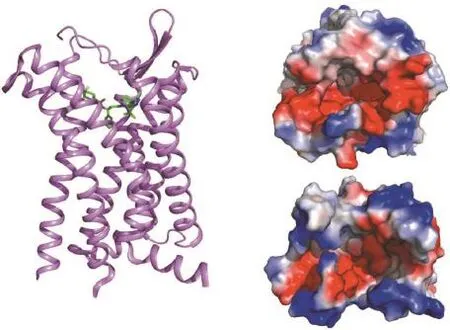
图1 CCR5和马拉维若的复合物晶体结构:(a)CCR5的晶体结构(紫色螺旋表示CCR5,绿色棍棒形结构表示马拉维若);(b) CXCR4胞外区表面电势图;(c)CCR5胞外区表面电势图(红色代表负电荷区域,蓝色代表正电荷区域)
2.2 嘌呤能受体P2Y1R的晶体结构
嘌呤能受体P2Y1R在人体内分布广泛,在心、脑、血管等组织中均有表达,其内源性激动剂主要是ADP和ATP。P2Y1R主要参与由ADP诱导的血小板聚集反应,与人体的各种严重的血栓性疾病密切相关,因此P2Y1R是极其重要的抗血栓药物靶点。2015年,我们解析了P2Y1R分别与核苷酸类拮抗剂MRS2500和非核苷酸类拮抗剂BPTU结合的复合物晶体结构,揭示了该受体蛋白与配体的特异性结合模式,对抗血栓药物研发具有重大意义[41]。
出乎意料的是,P2Y1R结构显示该受体具有完全不同的两个配体结合位点。其中,核苷酸类拮抗剂MRS2500的结合位点位于P2Y1R的跨膜螺旋区内部,其配体结合口袋由受体的N末端、第二、第六、第七跨膜螺旋以及第二胞外环构成(图2(b)),受体与MRS2500间的相互作用力主要是氢键和盐桥作用。P2Y1R与另外一种嘌呤能受体P2Y12R均以ADP为天然配体,但是比较这两种受体的晶体结构时发现两者与各自核苷酸类配体的结合位点和作用方式有很大差异[41-42]。P2Y1R结构中核苷酸类配体MRS2500的腺嘌呤环靠近受体的第六、第七跨膜螺旋,结合口袋位置更靠近胞外区,而P2Y12R结构中核苷酸类配体2MeSADP的腺嘌呤环深入配体结合口袋内部,与受体的第三、第四跨膜螺旋形成疏水作用[41-42]。这一发现充分体现了GPCR对信号识别机制的多样性。
更引起结构科学家们关注的是,非核苷酸类拮抗剂BPTU的结合位点位于P2Y1R跨膜螺旋的外表面,与脂分子双层相互接触(图2(a)),而在以往解析的GPCR结构中配体分子全部位于第七次跨膜螺旋区内部。BPTU是首个被发现的位于受体分子常规配体结合口袋以外区域的高选择性GPCR配体分子。BPTU的结合口袋由受体第一、第二、第三跨膜螺旋和第一胞外环上的疏水氨基酸构成,主要通过疏水作用与BPTU相互作用,唯一的极性作用是BPTU分子中尿素基团上的两个N原子与P2Y1R的氨基酸L102形成的两个氢键。BPTU的结合方式说明BPTU是P2Y1R的变构调节剂,而进一步的配体结合实验表明,BPTU可通过稳定受体的非活性构象提高激动剂的解离速度,从而达到对P2Y1R进行变构调节的目的。P2Y1R与BPTU的复合物结构首次揭示了GPCR在常规配体结构口袋以外区域的新型变构配体结合位点,有助于进一步全面了解GPCR受体的特异性配体结合模式,并为未来开展GPCR药物研发指明了新的方向。
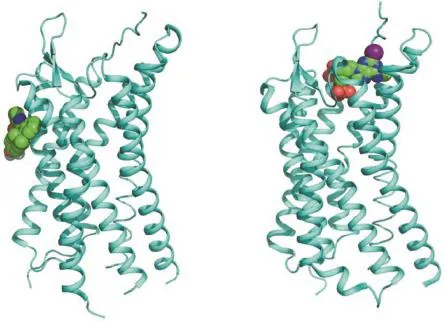
图2 P2Y1R 的两个结合口袋。(a)P2Y1R 与BPTU的复合物晶体结构(蓝色螺旋代表P2Y1R,球形结构代表BPTU);(b)P2Y1R 与MRS2500的复合物晶体结构(蓝色螺旋代表P2Y1R,球形结构代表MRS2500)
以往的研究表明,GPCR受体分子从非活性状态向活性状态转变时,第五至第七跨膜螺旋发生了较大的构象变化,而第一至四跨膜螺旋处于相对稳定的构象状态[43-44],因此人们一度认为GPCR的第五至第七跨膜螺旋在受体激活过程中发挥关键作用。在P2Y1R结构中,MRS2500结合在受体的跨膜螺旋区内,通过限制P2Y1R的第六、第七跨膜螺旋的构象来抑制受体激活。然而,在与BPTU结合的P2Y1R结构中,BPTU与第五至第七跨膜螺旋并无相互作用,而是通过阻碍第二、第三跨膜螺旋的构象变化抑制受体的激活。这一发现说明,虽然GPCR的第一至第四跨膜螺旋与第五至第七跨膜螺旋相比,在受体激活过程中发生的构象变化较小,但两个结构域对于受体的激活同等重要。这为我们进一步全面了解GPCR的激活机制提供了新的依据。
3 展望
在过去的几十年,GPCR结构信息的缺乏限制了科学家们对GPCR的功能研究和药物研发。近年来,GPCR的结构研究得到了跨越式的发展,使我们对这一重要的受体超家族有了较为深入的了解。随着更多GPCR精细结构逐渐展现在人们面前,GPCR对细胞信号的识别、传导和调控机制将得到更为全面的理解和认识。同时,GPCR的三维结构也有助于相关药物设计和研发,对于人类疾病的治疗具有重要意义。然而,目前GPCR的生物学研究还有许多问题尚未解决,因此GPCR的结构生物学研究仍然需要我们的不懈努力。
(2016年5月10日收稿)
[1] AUDET M, BOUVIER M. Restructuring G-protein-coupled receptor activation [J]. Cell, 2012, 151(1): 14-23.
[2] WISE A, GEARING K, REES S. Target validation of G-protein coupled receptors [J]. Drug Discovery Today, 2002, 7(4): 235-246.
[3] FREDRIKSSON R, LAGERSTROM M C, LUNDIN L G, et al. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints [J]. Molecular Pharmacology, 2003, 63(6): 1256-1272.
[4] OKADA T, LE TRONG I, FOX B A, et al. X-Ray diffraction analysis of three-dimensional crystals of bovine rhodopsin obtained from mixed micelles [J]. Journal of Structural Biology, 2000, 130(1): 73-80.
[5] STEVENS R C, CHEREZOV V, KATRITCH V, et al. The GPCR Network: a large-scale collaboration to determine human GPCR structure and function [J]. Nature Reviews Drug Discovery, 2013, 12(1): 25-34.
[6] ZHAO Q, WU B L. Ice breaking in GPCR structural biology [J]. Acta Pharmacologica Sinica, 2012, 33(3): 324-334.
[7] KEMPF J, SNOOK L A, VONESCH J L, et al. Expression of the human mu opioid receptor in a stable Sf9 cell line [J]. Journal of Biotechnology, 2002, 95(2): 181-187.
[8] KAUSHAL S, RIDGE K D, KHORANA H G. Structure and function in rhodopsin: the role of asparagine-linked glycosylation [J]. Proceedings of the National Academy of Sciences of the United States of America, 1994, 91(9): 4024-4028.
[9] O'MALLEY M A, MANCINI J D, YOUNG C L, et al. Progress toward heterologous expression of active G-protein-coupled receptors inSaccharomyces cerevisiae: Linking cellular stress response with translocation and trafficking [J]. Protein Science : A Publication of the Protein Society, 2009, 18(11): 2356-2370.
[10] ZHANG R, KIM T K, QIAO Z H, et al. Biochemical and mass spectrometric characterization of the human CB2 cannabinoid receptor expressed inPichia pastoris—importance of correct processing of the N-terminus [J]. Protein Expression and Purification, 2007, 55(2): 225-235.
[11] BOCKENHAUER S, FURSTENBERG A, YAO X J, et al. Conformational dynamics of single G protein-coupled receptors in solution [J]. The Journal of Physical Chemistry B, 2011, 115(45): 13328-13338.
[12] THOMPSON A A, LIU J J, CHUN E, et al. GPCR stabilization using the bicelle-like architecture of mixed sterol-detergent micelles [J]. Methods, 2011, 55(4): 310-317.
[13] PATEL R C, KUMAR U, LAMB D C, et al. Ligand binding to somatostatin receptors induces receptor-specific oligomer formation in live cells [J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(5): 3294-3299.
[14] RASMUSSEN S G, CHOI H J, ROSENBAUM D M, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor [J]. Nature, 2007, 450(7168): 383-387.
[15] SIU F Y, HE M, DE GRAAF C, et al. Structure of the human glucagon class B G-protein-coupled receptor [J]. Nature, 2013, 499(7459): 444-449.
[16] CHUN E, THOMPSON A A, LIU W, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors [J]. Structure, 2012, 20(6): 967-976.
[17] ROTH C B, HANSON M A, STEVENS R C. Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure [J]. Journal of Molecular Biology, 2008, 376(5): 1305-1319.
[18] LEBON G, BENNETT K, JAZAYERI A, et al. Thermostabilisation of an agonist-bound conformation of the human adenosine A(2A) receptor [J]. Journal of Molecular Biology, 2011, 409(3): 298-310.
[19] STEYAERT J, KOBILKA B K. Nanobody stabilization of G proteincoupled receptor conformational states [J]. Current Opinion in Structural Biology, 2011, 21(4): 567-572.
[20] LANDAU E M, ROSENBUSCH J P. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins [J]. Proceedings of the National Academy of Sciences of the United States of America, 1996, 93(25): 14532-14535.
[21] CHEREZOV V, ROSENBAUM D M, HANSON M A, et al. Highresolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor [J]. Science, 2007, 318(5854): 1258-1265.
[22] RASMUSSEN S G, DEVREE B T, ZOU Y, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex [J]. Nature, 2011, 477(7366): 549-555.
[23] KANG Y, ZHOU X E, GAO X, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser [J]. Nature, 2015, 523(7562): 561-567.
[24] NIBBS R J, YANG J, LANDAU N R, et al. LD78beta, a non-allelic variant of human MIP-1alpha (LD78alpha), has enhanced receptor interactions and potent HIV suppressive activity [J]. The Journal of Biological Chemistry, 1999, 274(25): 17478-17483.
[25] COMBADIERE C, AHUJA S K, TIFFANY H L, et al. Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1(alpha), MIP-1(beta), and RANTES [J]. Journal of Leukocyte Biology, 1996, 60(1): 147-152.
[26] GONG W, HOWARD O M, TURPIN J A, et al. Monocyte chemotactic protein-2 activates CCR5 and blocks CD4/CCR5-mediated HIV-1entry/replication [J]. The Journal of Biological Chemistry, 1998, 273(8): 4289-4292.
[27] CASTELLINO F, HUANG A Y, ALTAN-BONNET G, et al. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction [J]. Nature, 2006, 440(7086): 890-895.
[28] DRAGIC T, LITWIN V, ALLAWAY G P, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5 [J]. Nature, 1996, 381(6584): 667-673.
[29] CARDOZO T, KIMURA T, PHILPOTT S, et al. Structural basis for coreceptor selectivity by the HIV type 1 V3 loop [J]. AIDS Research and Human Retroviruses, 2007, 23(3): 415-426.
[30] WU B, CHIEN E Y, MOL C D, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists [J]. Science, 2010, 330(6007): 1066-1071.
[31] TAN Q, ZHU Y, LI J, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex [J]. Science, 2013, 341(6152): 1387-1390.
[32] BALASUBRAMANIAN C, CHILLEMI G, ABBATE I, et al. Importance of V3 loop flexibility and net charge in the context of coreceptor recognition. A molecular dynamics study on HIV gp120 [J]. Journal of Biomolecular Structure & Dynamics, 2012, 29(5): 1-13.
[33] XIANG S H, PACHECO B, BOWDER D, et al. Characterization of a dual-tropic human immunodeficiency virus (HIV-1) strain derived from the prototypical X4 isolate HXBc2 [J]. Virology, 2013, 438(1): 5-13.
[34] BISCONE M J, MIAMIDIAN J L, MUCHIRI J M, et al. Functional impact of HIV coreceptor-binding site mutations [J]. Virology, 2006, 351(1): 226-236.
[35] FDA notifications. Maraviroc approved as a CCR5 co-receptor antagonist [J]. AIDS Alert, 2007, 22(9): 103.
[36] WATSON C, JENKINSON S, KAZMIERSKI W, et al. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor [J]. Molecular Pharmacology, 2005, 67(4): 1268-1282.
[37] GARCIA-PEREZ J, RUEDA P, ALCAMI J, et al. Allosteric model of maraviroc binding to CC chemokine receptor 5 (CCR5) [J]. The Journal of Biological Chemistry, 2011, 286(38): 33409-33421.
[38] DUMA L, HAUSSINGER D, ROGOWSKI M, et al. Recognition of RANTES by extracellular parts of the CCR5 receptor [J]. Journal of Molecular Biology, 2007, 365(4): 1063-1075.
[39] SCHNUR E, KESSLER N, ZHERDEV Y, et al. NMR mapping of RANTES surfaces interacting with CCR5 using linked extracellular domains [J]. The FEBS Journal, 2013, 280(9): 2068-2084.
[40] KATRITCH V, CHEREZOV V, STEVENS R C. Structure-function of the G protein-coupled receptor superfamily [J]. Annual Review of Pharmacology and Toxicology, 2013, 53: 531-556.
[41] ZHANG D, GAO Z G, ZHANG K, et al. Two disparate ligand-binding sites in the human P2Y1 receptor [J]. Nature, 2015, 520(7547): 317-321.
[42] ZHANG J, ZHANG K, GAO Z G, et al. Agonist-bound structure of the human P2Y12 receptor [J]. Nature, 2014, 509(7498): 119-122.
[43] PARK J H, SCHEERER P, HOFMANN K P, et al. Crystal structure of the ligand-free G-protein-coupled receptor opsin [J]. Nature, 2008, 454(7201): 183-187.
[44] XU F, WU H, KATRITCH V, et al. Structure of an agonist-bound human A2A adenosine receptor [J]. Science, 2011, 332(6027): 322-327.
(编辑:段艳芳)
Structural studies of G protein-coupled receptors
ZHANG Haonan, WU Beili
CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
G protein-coupled receptors (GPCRs) play key roles in cell signal transduction. GPCR structures are important for understanding structure-functional relationship of the GPCR superfamily. Before the year 2000, the high-resolution GPCR structure is one of the major obstacles in structural biology field. In recent years, the structural studies of GPCRs have been developed remarkably. Here, we briefly summarize the methods used in GPCR structure determination, and also take the CCR5 and P2Y1R structures as examples to discuss the significance of GPCR structures on functional studies and drug discovery.
G protein-coupled receptor, crystal structure, CCR5, P2Y1R
10.3969/j.issn.0253-9608.2016.03.006
†通信作者,E-mail: beiliwu@simm.ac.cn
猜你喜欢
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
心肺血管病杂志(2019年1期)2019-04-22 01:12:00
教学考试(高考生物)(2017年4期)2017-12-13 09:02:41
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
应用化工(2014年7期)2014-08-09 09:20:23
火炸药学报(2014年3期)2014-03-20 13:17:39
无机化学学报(2014年5期)2014-02-28 17:31:40
华东理工大学学报(自然科学版)(2014年2期)2014-02-27 13:48:43
郑州大学学报(理学版)(2013年2期)2013-03-11 20:30:30
