
香附药材HPLC指纹图谱研究
2016-03-03 20:28蒋莉娟皮胜玲方铁铮姚江雄
中国当代医药 2016年1期
蒋莉娟+++皮胜玲++方铁铮+++姚江雄++++许招懂++++林丽美

[摘要] 目的 建立香附药材HPLC指纹图谱,结合化学计量手段,对多批次药材进行质量控制。 方法 以Kromasil 100-C18(250 mm×4.6 mm,5 μm)为色谱柱,以甲醇(A)-水(B)为流动相,梯度洗脱,流速为1.0 ml/min,检测波长为254 nm;柱温25℃,对10批药材样品进行相似度分析、聚类分析和主成分分析。 结果 建立了香附药材专属性的HPLC指纹图谱,标示了16个共有指纹峰,10批样品分为3大类。 结论 将HPLC指纹图谱与化学计量手段相结合,可有效对香附药材进行真伪鉴别和质量评价,为提高其整体质量控制提供参考。
[关键词] 香附;高效液相色谱法;指纹图谱;相似度评价;聚类分析;主成分分析
[中图分类号] R927.11 [文献标识码] A [文章编号] 1674-4721(2016)01(a)-0004-04
S 香附为莎草科植物莎草(Cyperus Rotundus L.)的干燥根茎,具有疏肝解郁、理气宽中、调经止痛的功效,用于治疗肝郁气滞所致的胸胁胀痛、疝气疼痛、乳房胀痛、脾胃气滞、脘腹痞闷、胀满疼痛、月经不调、经闭痛经之症,为妇科常用药[1]。《本草纲目》称之为“气病之总司,女科之主帅”,主产于山东、浙江、湖南、河南等地,以山东为道地药材。香附主要含挥发油类成分,此外还有萜类、黄酮类、酚类、生物碱等化合物[2]。现代药理研究显示,香附具有抗炎、抗血小板、降血糖、保护神经系统、抗过敏等作用[3]。指纹图谱是基于对中药物质群整体作用的认识,借助于波谱和色谱等技术获得中药化学成分的光谱或色谱图,是实现鉴别中药真实性、评价质量一致性和产品稳定性的可行模式,具有信息量大、特征性强、整体性和模糊性等特点[4]。本研究采用高效液相色谱(high performance liquid chromatography,HPLC)对10批香附药材进行研究并建立指纹图谱,旨在为香附药材的质量控制提供科学依据。
1 仪器与材料
1.1 仪器
KQ-100B型超声波清洗器(昆山超声仪器有限公司);T-214型电子分析天平(Denver公司);Waters e2695高效液相色谱系统,Empower工作站,含四元梯度泵、自动进样器、紫外检测器(Waters公司)。
1.2 材料
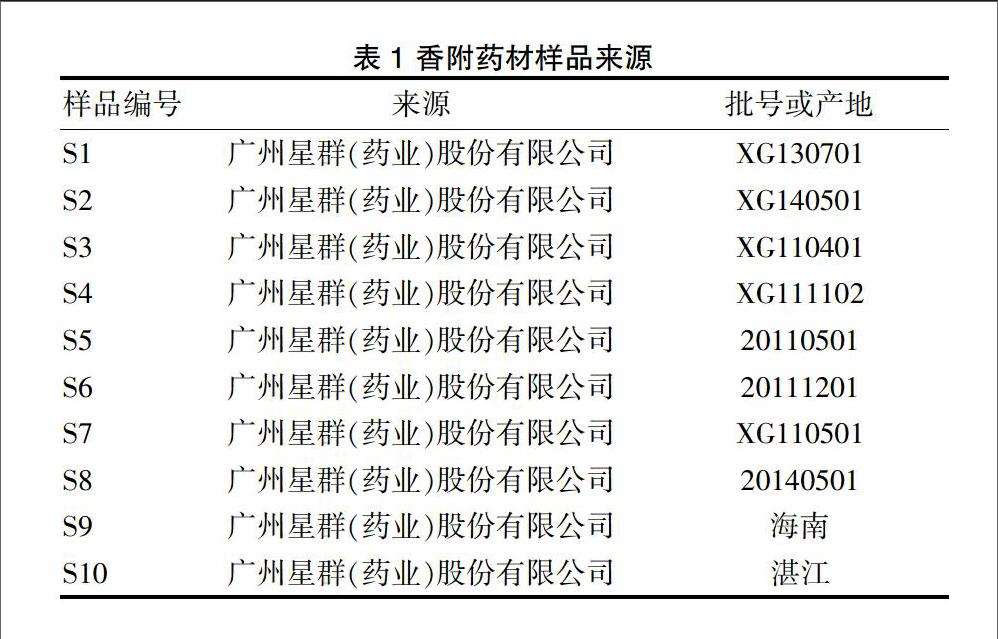
甲醇(Honeywell公司,色谱纯);水为超纯水(实验室制备)。α-香附酮对照品(批号:110748200507)购自中国药品生物制品检定所,供含量测定用。10份不同批次或产地的香附药材样品均由广州星群(药业)股份有限公司提供,经湖南中医药大学药学院药用植物教研室王智老师鉴定为莎草科植物莎草的干燥根茎,具体信息见表1。
表1 香附药材样品来源
2 方法与结果
2.1 色谱条件
色谱柱:Kromasil 100-C18(250 mm×4.6 mm,5 μm);流动相:甲醇(A)-水(B)为流动相系统梯度洗脱(0~45 min,40%→80%A;45~60 min,80%A);流速:1.0 ml/min;检测波长:254 nm;柱温:25℃;进样体积:10 μl。
2.2 对照品溶液的制备
精密称取α-香附酮对照品适量,加甲醇溶解定容至10 ml,得到浓度为0.0261 mg/ml的对照品溶液。
2.3 供试品溶液的制备
取香附药材粉末(过60目筛)约1 g,精密称定,置100 ml具塞锥形瓶中,精密加入甲醇10 ml,密塞,称重,超声处理(功率160 W,频率59 kHz)30 min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,0.22 μm微孔滤膜滤过,取续滤液,即得供试品溶液。
2.4 方法学考察
2.4.1 精密度试验 取同一批(XG140501)药材粉末,按“2.3”项下方法制备供试品溶液,按“2.1”项下色谱条件,连续进样6次,测定指纹图谱。以7号峰的保留时间和峰面积为参照,计算样品中所含16个共有峰的相对保留时间和相对峰面积。各共有峰相对保留时间的RSD为0.003%~0.130%,相对峰面积的RSD为0.15%~2.10%,提示仪器的精密度良好。
2.4.2 重复性试验 取同一批(20111201)药材粉末,按“2.3”项下方法平行制备6份供试品溶液,按“2.1”项下色谱条件,分别进样6次,测定指纹图谱。考察样品中所含16个共有峰的相对保留时间和相对峰面积,各共有峰相对保留时间的RSD为0.01%~0.48%,相对峰面积的RSD为0.01%~2.77%,提示方法的重复性良好。
2.4.3 稳定性试验 取1份(20111201)药材粉末,按“2.3”项下方法制备供试品溶液,分别于0、4、8、12、15、24 h测定指纹图谱,考察样品中所含16个共有峰的相对保留时间和相对峰面积,各共有峰相对保留时间的RSD为0.10%~0.63%,相对峰面积的RSD为0.33%~2.74%,提示香附药材样品溶液在24 h内稳定性良好。
2.5 指纹图谱的建立
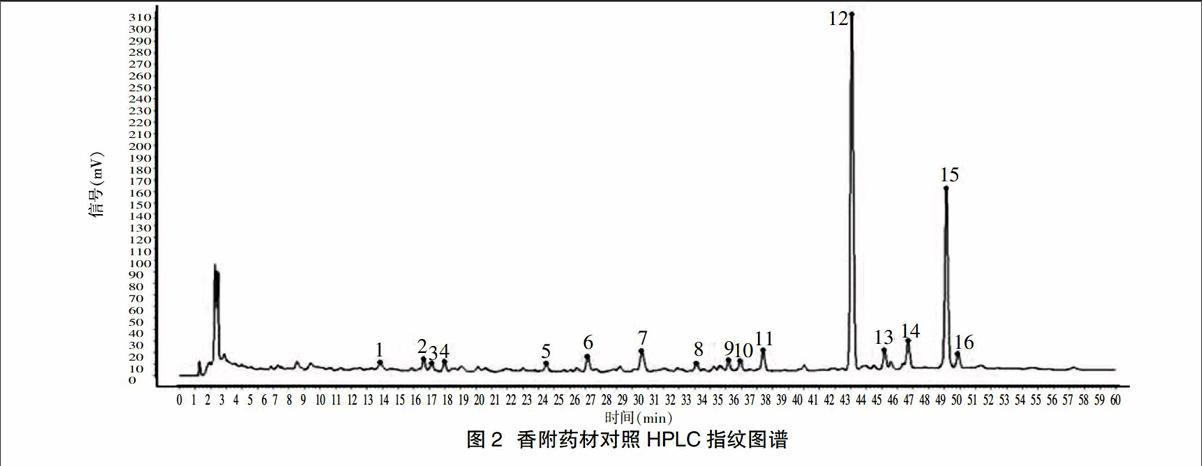
2.5.1 指纹图谱共有模式的建立 取10批药材粉末,按“2.3”项下方法制备供试品溶液,分别进样10 μl,记录HPLC色谱图(图1)。采用国家药典委员会中药色谱指纹图谱相似度评价系统(2.0版)进行分析,生成共有模式指纹图谱(图2),其中以峰7为参照物,其余各共有峰与峰7的相对保留时间与相对峰面积见表2、表3。
2.5.2 共有峰的确定 10批香附生成的对照指纹图谱标定了16个共有峰,具体见图2。对照品溶液的色谱图见图3,经与对照品对照,指认15号峰为α-香附酮。
2.5.3 指纹图谱的相似度评价 上述10批香附药材相似度计算结果依次为0.999、0.988、0.966、0.996、0.981、 0.771、0.997、0.913、0.996、0.975,除批次为20111201的香附药材相似度为0.771外,其余批次相似度均>0.9,提示香附药材样品具有良好相似性。
2.6 聚类分析
将10批香附药材HPLC指纹图谱中16个色谱峰的峰面积与样品号建立矩阵,导入SPSS软件,采用组间联接法,以欧氏距离的平方为测度,对数据进行标准化转换,得到香附药材的聚类分析图(图4)。结果显示,10批样品可分为三大类,S1、S2、S3、S4、S6、S7、S8、S9聚为一类,S5、S10分别单独聚为一类。聚类结果显示,药材各批次间成分总体差异较小。
2.7 主成分分析
采用SPSS软件对10批香附药材样品进行主成分分析,具体见图5。结果显示,S1、S2、S3、S4、S6、S7、S8、S9集中分布在一个区域,各样品相互之间距离较近;S5、S10与其他样品相距较远,样品可分为3大类。该图直接反映了各批次样品之间的差异大小,与聚类分析结果一致。
3 讨论
3.1 色谱条件的优化
本实验比较了245、254、265、300和320 nm波长下的色谱图[5-9],结果在254 nm检测条件下出峰较多,各峰吸收均匀,峰形较好,故选择254 nm作为检测波长。本实验考察了乙腈-0.5%冰乙酸水、乙腈-水、甲醇-水、甲醇-0.5%冰乙酸水不同流动相体系[10-14],结果显示甲醇-水能对样品中各成分实现较好的分离,因此流动相组成选择甲醇-水;还考察了25、30、35℃柱温条件下各色谱峰的分离情况,结果显示,不同柱温下色谱峰略有区别,当温度升高时,部分峰发生相对位移与合并,使分离度降低,因此选择柱温25℃;此外,本实验比较了Kromasil 100-5C18(250 mm×4.6 mm)、LiChrospher■100 RP-18 endcapped(5 μm)Hibar■RT 250-4.6、Athena C18120A(250 mm×4.6 mm,5 μm)、Agilent Eclipse XDB-C18(250 mm×4.6 mm,5 μm)4种不同型号的色谱柱,结果显示,Kromasil柱各色谱峰的分离度、峰形较其他色谱柱好,因此本实验选择Kromasil色谱柱。
3.2 提取方法的考察
本实验分别对提取溶剂(甲醇、50%甲醇、75%甲醇、乙醇、50%乙醇、75%乙醇)、提取方式(超声、加热回流)、提取时间(15、30、45、60 min)及提取体积(10、20、25、30 ml)进行了考察[15-18],基于出峰数目和峰面积最终选用甲醇超声提取30 min,提取体积10 ml,该方法提取较为完全,提取效率最高。
3.3 相似度结果分析
10批不同批次或产地的香附药材HPLC指纹图谱相似度较高,除批次20111201的香附药材相似度为0.771外,其余批次的相似度均>0.9,提示药材主要成分构成基本一致,药材质量稳定。主成分分析与聚类分析的结果相互佐证,结果一致,药材样品可聚为三类,S3和S10与其他样品相距较远,提示三类药材质量存在一定差异。
3.4 实验结果分析
不同批次或产地的香附药材HPLC指纹图谱既有共性又存在差异,主要色谱峰的整体面貌基本一致,但各峰峰面积有所不同,这说明环境因素会对药材质量产生一定影响,是否与生长环境、土壤条件等因素有关有待进一步考察[19-21]。本实验应用指纹图谱和聚类分析、主成分分析相结合的方法进行研究,能够更全面、准确地判别药材的真伪与优劣,可有效用于香附药材的质量控制与鉴定。
[参考文献]
[1] 国家药典委员会.中华人民共和国药典一部[M].北京:中国医药科技出版社,2010:241.
[2] 周中流,尹文清,张华林,等.香附化学成分研究[J].中草药,2013,44(10):1226-1230.
[3] 徐晓婷,邓志鹏,仲浩,等.香附化学成分及药理作用研究进展[J].齐鲁药事,2012,31(8):473-475.
[4] 李强,杜思邈,张忠亮,等.中药指纹图谱技术进展及未来发展方向展望[J].中草药,2013,44(22):3095-3102.
[5] 卢君蓉,李文兵,王世宇,等.香附醋制前后香附烯酮、圆柚酮和α-香附酮的含量比较[J].中国实验方剂学杂志,2014,20(20):24-27.
[6] 王世宇,李文兵,卢君蓉,等.HPLC法同时测定不同产地香附药材中香附烯酮、圆柚酮和α-香附酮[J].中成药,2015,37(3):588-591.
[7] 季宁平,卢君蓉,李文兵,等.不同醋制方法对香附中指标成分含量的影响[J].中国实验方剂学杂志,2015,21(7):5-7.
[8] 徐媛,张文娟,王庆伟,等.香附不同炮制品HPLC指纹图谱研究[J].中国医药导报,2011,8(31):29-32.
[9] 高景莘,夏厚林,吴远波.香附药材HPLC指纹图谱研究及相似度评价[J].中国中医药咨讯,2010,2(14):240-241.
[10] 赵新慧,宿树兰,段金廒,等.香附药材质量相关性分析研究[J].药物分析杂志,2008,28(2):187-192.
[11] 童黄锦,白发平,汪小莉,等.不同产地白芍药材的指纹图谱研究[J].中医学报,2014,29(9):1326-1329.
[12] 徐昌艳,张丽艳,汪毅,等.苗药矮地茶的HPLC指纹图谱研究[J].中药材,2014,37(9):1570-1573.
[13] 孙莲,严寒信,贾晓琴.HPLC同时测定芜菁子药材中槲皮素和山奈酚含量及其指纹图谱的研究[J].中国现代应用药学,2014,31(11):1347-1351.
[14] 闵会,吴健,胡江宁,等.不同贝母的指纹图谱鉴别研究[J].安徽农业科学,2014,42(34):12051-12052.
[15] 魏刚,顺庆生,戴亚峰,等.霍山石斛HPLC特征图谱研究[J].中成药,2014,36(12):2642-2644.
[16] 焦豪妍,王英,陈丽莉,等.广东道地药材何首乌HPLC指纹图谱研究[J].中国药业,2014,23(23):58-61.
[17] 何卓琳,王帅,孟宪生,等.基于指纹图谱的益母草胶囊质量控制方法研究[J].中国当代医药,2013,20(32):7-9.
[18] 毛健,刘振杰.余甘子叶药材的 HPLC 指纹图谱研究[J].中国当代医药,2014,21(17):11-14.
[19] 王黎,高苏亚,李华.不同产地葛根的HPLC指纹图谱及质量评价研究[J].安徽农业科学,2011,39(18):10782-10784.
[20] 甘秀海,梁志远,姜金仲,等.冷水花药材的HPLC指纹图谱研究及指标成分含量测定[J].中药材,2015,38(2):275-278.
[21] 孙冬梅,丘思兰,李素梅,等.银杏叶药材HPLC指纹图谱研究[J].辽宁中医杂志,2015,42(2):366-368.
(收稿日期:2015-08-12 本文编辑:祁海文)
猜你喜欢
中国中药杂志(2016年22期)2017-02-13
中国民族民间医药·上半月(2016年12期)2017-01-11
中国医药导报(2016年30期)2016-12-28
中国民族民间医药·上半月(2016年11期)2016-12-26
中国中药杂志(2016年20期)2016-11-19
中国民族民间医药·上半月(2016年10期)2016-11-19
妇女生活(2016年9期)2016-09-08
天津农业科学(2015年5期)2015-05-30
中成药(2015年8期)2015-01-18
中国中医药现代远程教育(2014年16期)2014-03-01
中国当代医药2016年1期