
CNTs-Al2O3联合增强镁基复合材料的制备与性能研究
2016-02-21 06:06于洋李海鹏程里宋晓庆秦剑坤
河北工业大学学报 2016年3期
于洋,李海鹏,程里,宋晓庆,秦剑坤
(河北工业大学材料科学与工程学院,天津300130)
CNTs-Al2O3联合增强镁基复合材料的制备与性能研究
于洋,李海鹏,程里,宋晓庆,秦剑坤
(河北工业大学材料科学与工程学院,天津300130)
采用化学气相沉积法,使用镍催化剂,在氧化铝(A l2O3)颗粒载体表面原位合成了碳纳米管(CNTs),制备出CNTs-A l2O3原位复合增强相;采用粉末冶金工艺制备了CNTs-A l2O3联合增强镁基复合材料.通过XRD、SEM、TEM等对CNTs-A l2O3原位复合增强相进行表征,结果表明:通过控制合成条件,在A l2O3颗粒表面可获得分散均匀、形态理想、石墨化程度高的CNTs,实现了CNTs与A l2O3颗粒的原位复合.对复合材料的组织和力学性能分析表明:A l2O3颗粒发挥了良好的“运载效果”,通过球磨工艺,使CNTs从A l2O3颗粒表面脱落并在镁基体中分散;同时,作为催化剂载体的A l2O3颗粒与CNTs对镁基体起到联合增强作用,使得复合材料力学性能明显提高.
碳纳米管;氧化铝;联合增强;镁基复合材料;化学气相沉积
0 引言
在装备制造轻量化的发展趋势下,镁基复合材料在航空航天、汽车、建材等领域的应用日益广泛.但镁基复合材料仍存在高温强度低、抗蠕变性和耐磨性差等不足,制约了其在装备制造领域的更广泛应用.自被发现以来,碳纳米管(CNTs)凭借其优异的机械和物理性能,被认为是高性能复合材料的理想增强相,关于CNTs增强镁基复合材料的研究也被深入开展.例如,Park等[1]采用挤压渗透法制备了CNTs/Si/Mg复合材料,CNTs/Si的加入减少了镁基复合材料的铸造缺陷并提高了镁基复合材料的抗拉强度.Han等[2]采用粉末冶金法制备了CNTs增强镁基复合材料,同原始基体相比,复合材料抗拉强度提升了22.7%,抗压强度提升了53%.张云鹤等[3]也采用粉末冶金法制备了CNTs增强镁基复合材料,所制备的复合材料组织致密,CNTs的加入使纯镁的硬度提高了18%.陈玉华等[4]以CNTs为增强相,采用搅拌摩擦加工法制备了镁基复合材料,CNTs的加入使复合区晶粒细化,复合材料显微硬度和抗拉强度都得到明显改善.但是,因CNTs长径比大、比表面能高,存在的较强范德华力使其在镁基复合材料中极易团聚,而传统方法难以解决其在镁基复合材料中的均匀分布问题[5-7].例如,简单的超声分散无法克服CNTs间的范德华力作用;高能球磨可能破坏CNTs的结构完整性,并导致氧化镁的形成[8];搅拌铸造法易导致CNTs的结构损伤和在块体材料中的偏聚[9].
为解决CNTs在金属基复合材料中的分散问题,研究人员已实现了在金属(如A l、Co、Cu等)粉末中CNTs的原位合成,达到了CNTs与金属基体粉末的“分子级混合”效果,使得复合材料的力学性能得到显著提高[10-14].然而,关于在镁基体中原位合成CNTs的尝试表明:尽管在较低温度下可获得CNTs,但镁和催化剂之间存在强烈的相互作用,导致CNTs的合成效果并不理想[15];即使严格控制化学气相沉积(CVD)过程以避免镁与催化剂的反应,仍不可逆地形成了不利于镁基复合材料性能的杂质和脆性MgO[16].因此,镁较低的熔点和活泼的化学性质,导致其并不是CNTs合成的理想载体,通过原位合成技术制备CNTs增强镁基复合材料仍存在难度.
基于此,结合原位合成技术的优点并避免杂质的形成,本文提出了CNTs-A l2O3联合增强镁基复合材料的制备方法.首先,使用镍催化剂,通过CVD法在微米Al2O3颗粒表面原位合成CNTs,制备CNTs-Al2O3原位复合增强相;然后,通过适度球磨使CNTs-A l2O3原位复合增强相分散到镁粉中.由于微米级颗粒间的范德华力较低,A l2O3颗粒易于在镁粉中分散;借助Al2O3颗粒的“运载”作用,生长在其表面短而直的CNTs在球磨阶段逐渐剥离并分散在镁粉中,从而间接达到了CNTs的分散效果.此外,与CNTs相似,A l2O3颗粒也是金属基复合材料的常用增强相[17],二者可实现对镁基体的联合增强.
1 实验方法
1.1 合成CNTs-A l2O3原位复合增强相
为使Ni催化剂均匀分布于Al2O3颗粒表面,采用沉积-沉淀法制备NiO/A l2O3催化剂前驱体,具体包括:将适量的Al2O3粉末(≤100m,纯度99%)加入浓度0.1mol L1的Ni(NO3)26H2O(纯度98%)去离子水溶液中(保证Ni/A l2O3的重量比为1∶11.5);在搅拌状态下逐滴滴加0.1mol L1的NaOH水溶液并持续搅拌0.5 h以上,静置24 h后经过滤、清洗得到Ni(OH)2/Al2O3二元胶体;将该胶体在N2气氛、450℃煅烧2 h,得到NiO/Al2O3催化剂前驱体;随后,关闭N2并通入H2(100m L/m in)将NiO/A l2O3还原为所需Ni/Al2O3催化剂.采用CVD法进行CNTs-Al2O3原位复合增强相的合成,合成条件为:合成温度700℃,合成时间分别为30min、60min和120m in,合成气氛为CH4(纯度99.9%,60m L/m in)与N2(纯度99.9%,420m L/min)的混合气体.复合增强相中CNTs的含量由式(1)确定

式中:WYield表示CNTs的合成产率,%;Wa表示CVD反应后复合粉末的质量,mg;Wb表示催化剂前驱体NiO/Al2O3的质量,mg.
1.2 制备CNTs-A l2O3/Mg复合材料
将纯镁粉(400目,纯度99.5%)和CNTs-A l2O3复合粉末放入行星式球磨机中,以400 r/m in的转速球磨24h.将球磨后的粉末放入直径20mm的冷压模具中,在600MPa压力下压制成圆柱状毛坯.然后将毛坯放入烧结炉中,在H2气氛、480℃条件下烧结1.5 h.将烧结后的毛坯在400℃预热后,采用H13热挤压模具对毛坯进行热挤压(挤压压力750MPa、挤压比16∶1),获得CNTs-A l2O3联合增强镁基复合材料.同时,采用相同的工艺制备纯镁块体、A l2O3/Mg复合材料和CNTs/Mg复合材料以进行对比.
1.3 结构表征与性能测试
使用HitachiS-4800型扫描电子显微镜(SEM)和TECNAIG2F20型场发射透射电子显微镜(TEM)观察CNTs-Al2O3复合粉末中CNTs的形貌结构.使用Bruke RFS 100/S型拉曼光谱(Raman)测试仪评价CNTs-A l2O3复合粉末中CNTs的石墨化程度(测试条件:激发源为Ar+激光,激发波长512nm).使用SDTQ-600差热分析仪(DSC-TGA/DTA)测定CNTs-Al2O3复合粉末中CNTs的热稳定性(测试条件:温度范围0~750℃,空气气氛,升温速率10℃/m in).利用DigitalM icrograph软件测量CNTs的石墨层片间距.使用EveroneMH-6型显微维氏硬度计测定复合材料的显微硬度(HV)(测试条件:载荷245.2mN、加载时间15 s).按照ASTM E8标准,使用CSS-44100型万能拉伸试验机测定复合材料的拉伸性能.对每种材料使用3个直径为2.5mm的圆柱形试样进行测试,拉伸速度为0.5mm/m in.
2 结果与讨论
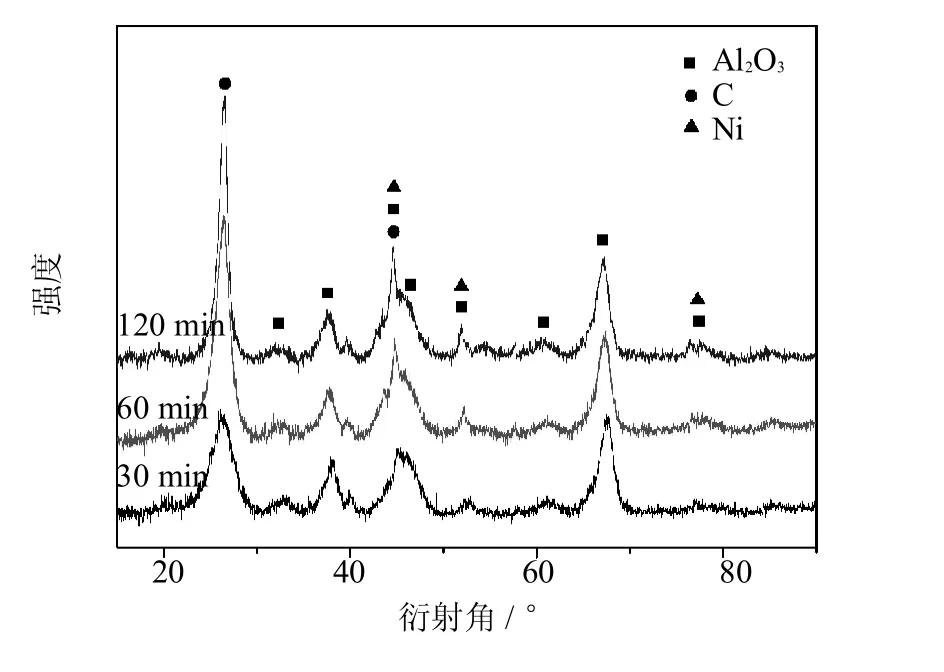
图1 不同合成时间所得CNTs-A l2O3原位复合粉末的XRD图Fig.1 XRD imagesof the CNTs-A l2O3composite powders fabricated atdifferentperiods

图2 氧化铝颗粒和不同合成时间所得CNTs-A l2O3原位复合粉末的SEM图Fig.2 SEM imagesof Al2O3particlesand CNTs-A l2O3composite powders fabricated atdifferentperiods
图1所示为不同合成时间(30m in、60m in和120m in)所得CNTs-A l2O3复合粉末的XRD图.由该图可见,不同合成时间的合成产物均包括A l2O3、C和Ni三相.位于2为26.2°和44.2°的衍射峰分别属于石墨(002)和(101)峰,表明合成产物中有晶化程度良好的CNTs生成;随着合成时间延长,位于26.2°附近的石墨衍射峰逐渐增强、半峰宽逐渐变窄,表明合成产物中CNTs的含量逐渐增加、尺寸逐渐增大.其余衍射峰分别对应Al2O3和Ni的特征峰.
图2所示为氧化铝颗粒和CNTs-A l2O3原位复合粉末的SEM图.由图2a)可见,原始氧化铝颗粒为规则球形,表面光洁,粒径约50m.当合成时间为30m in时,如图2b)所示,大量短小的CNTs均匀覆盖了A l2O3颗粒表面,CNTs长度在200~300nm之间,产率仅为4.1%.随合成时间延长,CNTs长度和产率都明显增加.如图2c)所示,合成时间60min所得到的CNTs平均长度达到1m,直径在20~25 nm,产率达到10.4%;CNTs与A l2O3载体结合良好,管体平直、光滑且未出现团聚缠绕现象,这有利于后续CNTs在镁基体中的分散及良好增强效果的发挥;合成产物中未明显观察到其它类型碳纳米结构,表明合成产物纯度较高.当合成时间为120 min时,由图2d)可见,除CNTs长度稍有增加外,合成时间的进一步延长并未显著改变CNTs的形貌和分散程度;CNTs合成产率为10.7%,与60m in的合成产率相近.
图3a)~图3c)所示为不同合成时间所得CNTs的TEM图.可发现,合成产物为典型多壁CNTs,且合成时间对CNTs形貌和结构有显著影响.合成时间为30 m in时,如图3a)所示,CNTs中空度较低,管壁中石墨层纹理不清晰,存在大量不完整的石墨层,呈现为短程有序而长程无序的状态,这表明其石墨化程度较低.随合成时间延长,CNTs的合成效果明显改善.如图3b)、3c)所示,当合成时间为60m in和120m in时,所合成的CNTs管壁光滑、平直,石墨层纹理清晰;使用Digital M icrograph软件测得石墨片层间距为0.341 nm,与理想石墨0.34 nm的片层间距非常接近,表明CNTs具有良好石墨化程度.对包覆在CNTs芯部黑色颗粒的EDX分析(如图3d)所示)表明,其为Ni催化剂颗粒;此外,TEM分析发现,几乎所有Ni催化剂颗粒都被包覆在CNTs的端部或芯部,从而避免了残留Ni催化剂对镁基复合材料性能的影响.由上述SEM和TEM分析可知,通过CVD法,借助Ni催化剂,在合成时间30~120m in内可在A l2O3颗粒表面均匀合成短而直的CNTs;氧化铝是CVD法合成CNTs的有效载体,对阻止CNTs团聚起到了重要作用[18],这些都有利于后续CNTs在镁基体中的均匀分散及其良好增强效果的发挥.
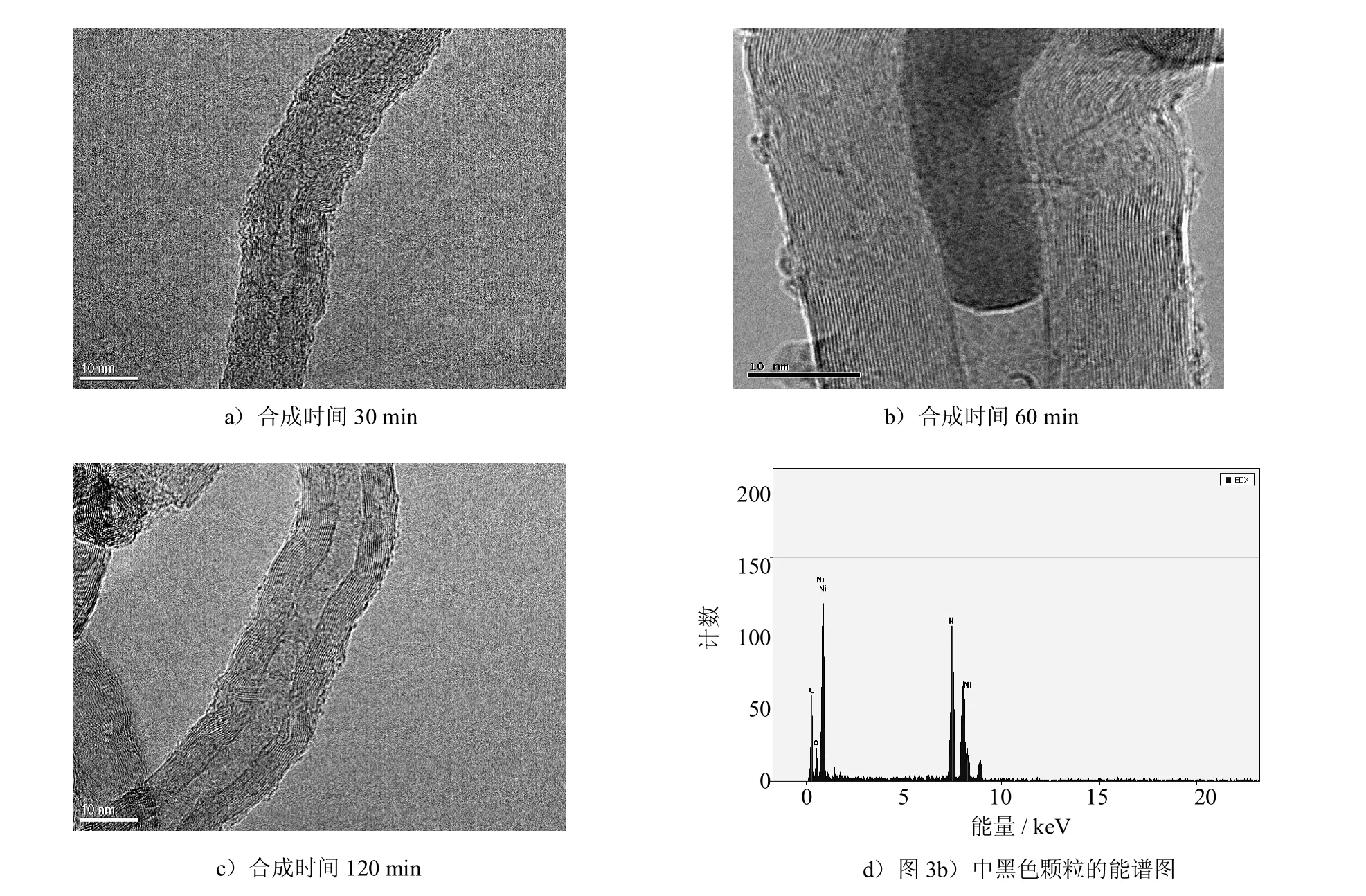
图3 不同合成时间所得CNTs的TEM分析Fig.3 TEM analysisof theCNTs fabricated atdifferentperiods
图4a)所示为不同合成时间所得CNTs-Al2O3原位复合粉末的TGA-DSC曲线,由此可评估所得CNTs的热氧化稳定性.由该图可见,所有样品均表现出相似的氧化行为和失重行为:在加热初始阶段,失重曲线无明显下降,说明CNTs石墨化程度高、非晶碳相存在较少[19];在500~680℃温度区间内,TGA曲线明显下降,这是由CNTs严重氧化失重所致.合成时间为30m in时,CNTs-A l2O3复合粉末的失重比率明显低于60min和120min,这是由于其CNTs含量较低所致,与XRD所反映出的产率趋势一致;样品的减重区间集中在440~600℃范围内,放热峰值出现温度较低(534℃)且半峰宽大,说明其石墨化程度较低,与TEM的观察结果相吻合.合成时间为60m in时,在460~680℃区间内CNTs发生剧烈氧化,在550℃时TGA曲线急剧下降、出现明显的失重台阶,653℃时出现半峰宽较窄的放热峰值,这说明该CNTs样品具有较高的热氧化稳定性和石墨化程度.进一步延长合成时间到120m in时,样品在460~640℃出现严重的氧化失重,在618℃出现放热峰值,均低于合成时间60m in时的对应温度,表明CNTs的热氧化稳定性较60m in时有所降低.由上述分析结果可知,合成时间60m in的CNTs热氧化温度高、抗氧化稳定性强,更适合作为镁基复合材料的增强相.图4b)所示CNTs的拉曼光谱分析结果进一步确认了合成时间对CNTs石墨化程度的影响,为优选镁基复合材料的CNTs-A l2O3增强相提供了依据.由该图可见,CNTs在1 340 cm1(D峰)和1590cm1(G峰)波谱位置出现2个强峰,具有典型多壁CNTs的拉曼光谱特征.合成时间60m in时CNTs的D峰较低、G峰较高(ID/IG=0.90),说明CNTs石墨化程度高、结构缺陷少.而合成时间为30m in和120m in时,CNTs的D峰较高、G峰较低,ID/IG分别为1.26和1.03,说明在合成产物中存在大量C悬键,石墨化程度较60min的合成效果明显降低.由此可见,合成时间为60m in时,A l2O3颗粒表面不仅CNTs合成产率较高,而且CNTs具有良好热氧化稳定性和较高石墨化程度,该条件下合成的CNTs-Al2O3原位复合粉末更适合作为镁基体的联合增强相.

图4 不同合成时间所得CNTs-A l2O3原位复合粉末的TGA-DSC曲线和Raman曲线Fig.4 TGA and Raman spectrum of CNTs-Al2O3composite powders fabricated atdifferentperiods
为比较不同增强相的增强效果,采用相同方法分别制备了A l2O3、CNTs和CNTs-A l2O3增强镁基复合材料.图5所示为3种镁基复合材料的显微硬度.由该图可见,3种复合材料的硬度均首先随增强相含量增加而增大;当增强相含量为4%(质量分数)时硬度达到峰值,CNTs-A l2O3联合增强的镁基复合材料硬度比纯镁提高了62.4%,且比单独加入相同含量A l2O3和CNTs的复合材料硬度分别提高了15.0%和7.8%;进一步提高增强相含量,3种复合材料的硬度均略有下降,但CNTs-Al2O3联合增强镁基复合材料硬度始终大于其它二者.由此可见,CNTs-A l2O3联合增强相提高镁基体硬度的效果更加明显.
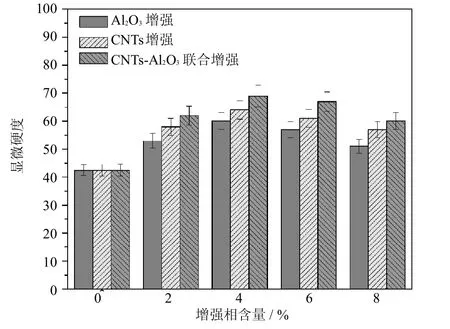
图5 A l2O3、CNTs和CNTs-A l2O3增强镁基复合材料的显微硬度Fig.5 M icrohardnessofMgmatrix composites reinforced by A l2O3, CNTsand CNTs-A l2O3
表1所示为根据拉伸试验获得的A l2O3、CNTs和CNTs-Al2O3增强镁基复合材料的抗拉强度和弹性模量.由表1可知,CNTs-Al2O3复合增强相有效改善了镁基体的力学性能,使得抗拉强度和弹性模量均有显著提高.纯镁试样的抗拉强度为189.4 MPa,加入CNTs-A l2O3复合增强相的镁基复合材料抗拉强度最低也达到214.1MPa(8%CNTs-A l2O3/Mg基复合材料);4%CNTs-A l2O3/Mg基复合材料的抗拉强度和弹性模量最高,分别比纯镁提高了21.3%和25.7%.对于单一CNTs和Al2O3增强相,虽然二者的加入使得复合材料抗拉强度和弹性模量均高于纯镁,但均低于同含量下CNTs-A l2O3/Mg的力学性能,例如,与CNTs-Al2O3/Mg相比,4% CNTs/Mg复合材料的抗拉强度和弹性模量分别下降了11.7%和18.9%.图6所示为4%CNTs-Al2O3/Mg复合材料拉伸断面的SEM图.由该图可见,在拉伸断面上均匀分布着被拔出的CNTs,在复合材料断裂过程中,上述CNTs可通过桥连、拔出等机制在镁基体中有效传递载荷、增大变形阻力,提高复合材料强度.
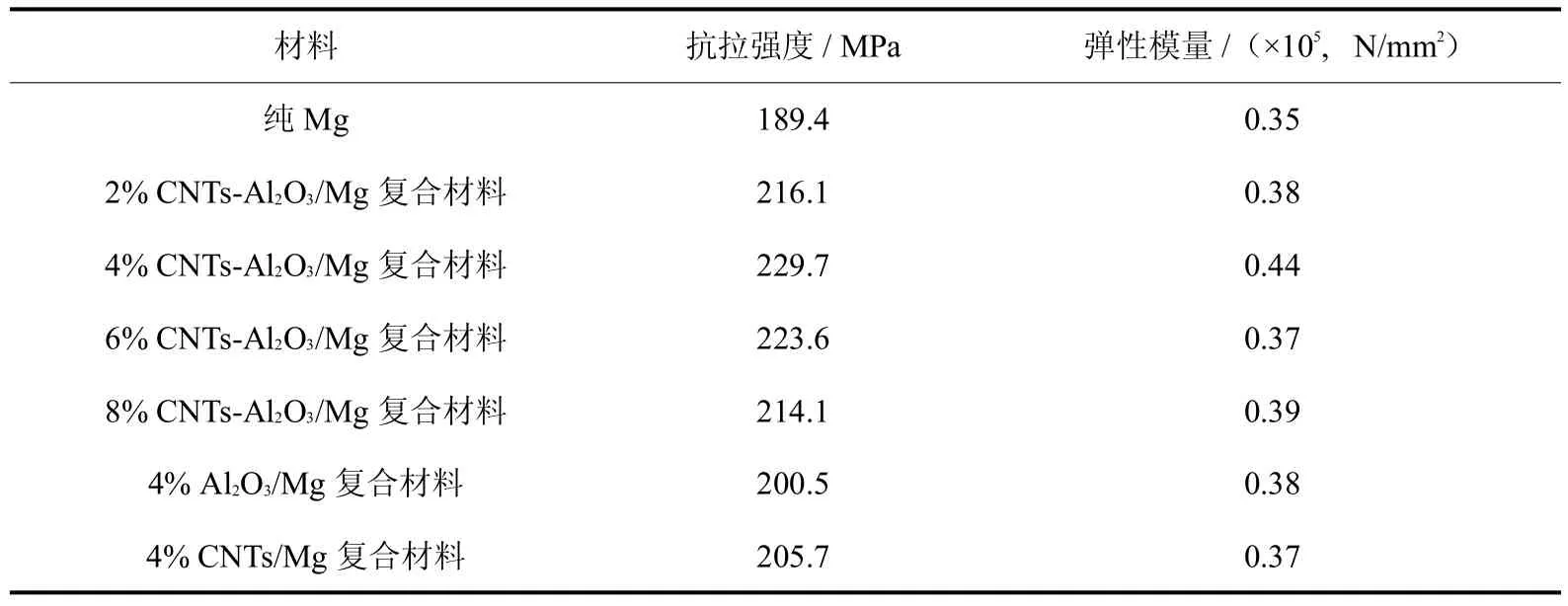
表1 A l2O3、CNTs和CNTs-A l2O3增强镁基复合材料的拉伸性能Tab.1 Tensile property ofMgmatrix composites reinforced by A l2O3,CNTsand CNTs-A l2O3

图6 CNTs-A l2O3/Mg复合材料拉伸断面的SEM图Fig.6 SEM imageof fracturalsurfaceof CNTs-A l2O3/Mg composite
复合材料的力学性能与增强相在基体中的分散情况以及增强相-基体之间的界面粘结状态密切相关[20-21].在CNTs-A l2O3/Mg复合材料制备过程中,通过适度球磨可使微米级A l2O3颗粒均匀分散在镁粉中;由于原位生长的CNTs与A l2O3颗粒之间仅存在较弱的机械结合,适度的球磨可以使CNTs逐渐从A l2O3颗粒表面剥离而不发生结构破坏,剥离下来的CNTs保持了结构完整性、避免了团聚,并与镁粉形成了良好的混合,从而保证了增强相在复合材料基体中的分散.在镁基体中分散均匀的CNTs可与镁基体形成良好的界面结合,起到钉扎晶界、阻碍晶粒长大、细化基体组织的作用,从而使位错在晶界处的迁移所受阻力更大,增加复合材料的变形阻力;CNTs还能够填充镁基体中的孔隙并在基体晶粒之间起到桥连作用,强化了基体晶粒之间的界面结合.此外,作为CNTs合成载体的A l2O3颗粒也是金属基复合材料的常用增强相,具有强度和硬度高、抗变形能力强的特点,可以阻碍镁基体塑性变形过程中的位错运动,从而起到强化作用.因此,CNTs-A l2O3的联合增强使得镁基体的力学性能得到提升,且增强效果优于单独的CNTs或A l2O3增强相.
3 结论
本文提出了一种利用CNTs-A l2O3原位复合结构联合增强镁基复合材料的制备方法,借助于CNTs合成载体A l2O3颗粒的输运作用,通过球磨工艺间接实现了CNTs在镁基体中的分散.采用CVD法、借助Ni催化剂,在A l2O3颗粒表面合成了分散均匀、热氧化稳定性和石墨化程度高的CNTs,获得了结构理想的CNTs-A l2O3原位复合结构.经CNTs-Al2O3的联合增强,镁基复合材料的力学性能得到改善,且增强效果优于单独的CNTs或A l2O3增强相.
[1]Park Y,Cho K,Park I,etal.Fabrication andmechanical propertiesofmagnesium matrix composite reinforced with Si coated carbon nanotubes [J].Proc Eng,2011,10(7):1446-1450.
[2]Han GQ,Wang ZH,Liu K,etal.SynthesisofCNT-reinforced AZ31magnesium alloy compositesw ithuniform ly distributed CNTs[J].MaterSci Eng A,2015,628:350-357.
[3]张云鹤,李庚,苗孟河,等.粉末冶金法碳纳米管增强镁基复合材料的微观组织及力学性能[J].复合材料学报,2013,30(增刊):102-106.
[4]陈玉华,刘东亚,谢吉林,等.搅拌摩擦加工法制备碳纳米管增强镁基复合材料的组织与性能[J].南昌航空大学学报(自然科学版),2015,29(1):49-54.
[5]周国华,曾效舒,袁秋红,等.消失模铸造法制备CNTs/ZM 5镁合金复合材料的研究[J].金属铸锻焊技术,2008,37(9):11-15.
[6]LiHP,Kang JL,HeCN,etal.Mechanicalpropertiesand interfacialanalysisof alum inum matrix composites reinforced by carbon nanotubesw ith diverse structures[J].Mater SciEng A,2013,577(9):120-124.
[7]Kondoh K,FukudaH,Umeda J,etal.M icrostructuralandmechanicalanalysisofcarbon nanotube reinforcedmagnesium alloy powdercomposites [J].Mater SciEng A,2010,527(16-17):4103-4108.
[8]王宝民,韩瑜,宋凯.碳纳米管分散性研究进展[J].材料导报,2012,26(4):23-30.
[9]周国华,曾效舒,袁秋红,等.铸造法制备CNTs/AM 60镁基复合材料的研究[J].铸造,2009,58(l):43-46.
[10]Cha SI,Kim KT,Arshad SN,etal.Field-em ission behavior of a carbon-nanotube-implanted Co nanocomposite fabricated from pearl-necklacestructured carbon nanotube/Co powders[J].Adv Mater,2006,18(5):553-558.
[11]ChaSI,Kim KT,Arshad SN,etal.Extraordinary strengtheningeffectof carbonnanotubesinmetal-matrix nanocompositesprocessed bymolecularlevelm ixing[J].Adv Mater,2005,17(11):1377-1381.
[12]HeCN,Zhao NQ,ShiCS,etal.Anapproach to obtaininghomogeneously dispersed carbonnanotubes in A lpowders forpreparing reinforced A lmatrix composites[J].Adv Mater,2007,19(8):1128-1132.
[13]Kang JL,Nash P,LiJJ,etal.Achievinghighly dispersed nanofibresathigh loading in carbonnanofibre-metalcomposites[J].Nanotechnology,2009,20(23):235607-235614.
[14]赵乃勤,马杰,师春生,等.原位化学气相沉积法制备碳纳米管增强金属基复合材料[J].金属热处理,2009,34(7):100-105.
[15]Kang JL,Li JJ,Zhao NQ,etal.Study ofMg powderascatalystcarrier for the carbon nanotubegrow th by CVD[J].JNanomater,2011,1155(10):938493-938498.
[16]Sun FJ,ShiCS,RheeKY,etal.In situ synthesisofCNTsinMg powderat low temperature for fabricating reinforcedMg composites[J].JAlloy Compd,2013,551:496-501.
[17]周玉成,魏世忠,王利敏,等.自生氧化铝颗粒增强铁基复合材料的制备[J].材料热处理技术,2011,40(22):100-102.
[18]He DL,BozlarM,Genestoux M,etal.Diameterand length-dependentself-organizationsofmulti-walled carbon nanotubeson sphericalalumina m icroparticles[J].Carbon,2010,48(4):1159-1170.
[19]Chen CM,Chen M,Peng YW,etal.High efficiencymicrowave digestion purification ofmulti-walled carbon nanotubessynthesized by thermal chem icalvapor deposition[J].Thin Solid Films,2006,498(1-2):202-205.
[20]Tham LM,Gupta M,Cheng L.Effectof lim itedmatrix-reinforcement interfacial reaction on enhancing themechanical propertiesof alum iniumsilicon carbide composites[J].ActaMater,2001,49(16):3243-3253.
[21]孙继兵,李国彬,安雅琴,等.原位自生成陶瓷相增强铝基复合材料的研究[J].河北工业大学学报,2000,29(6):93-96.
[责任编辑 田丰 夏红梅]
Preparation and propertiesof CNTs-Al2O3jointly reinforced magnesium matrix composites
YU Yang,LIHaipeng,CHENG Li,SONG Xiaoqing,QIN Jiankun
(SchoolofMaterials Scienceand Engineering,HebeiUniversity of Technology,Tianjin 300130,China)
Carbon nanotubes(CNTs)were synthesized in-situ over A l2O3particlesusing Nicatalystsby chem icalvapor deposition,achieving the joint reinforcementof CNTs-A l2O3.And then theMgmatrix composites reinforced by CNTs-A l2O3jointreinforcementwere fabricated by choosing the powdermetallurgy process.The structuralcharactersofCNTs-A l2O3joint reinforcementwere investigated by XRD,SEM,TEM,etc.The results show that,by controlling the synthesis conditions,the CNTs synthesized over A l2O3particles possessed homogeneous dispersion,idealmorphology and high degree ofgraphitization.When being used to reinforce theMgmatrix composites,due to the vehicle carrying effectofA l2O3particles,CNTswerepeeled off from A l2O3particlesand dispersed inMgmatrix duringballm illing process. Combining w ith the reinforcing effectof A l2O3particles,the synergistic reinforcementof CNTs and A l2O3wasobtained inMgmatrix composite.Consequently,theMgmatrix composites reinforced by CNTs-A l2O3m ixtureexhibited improved mechanicalproperties.
carbon nanotubes;alum ina;jointreinforcement;magnesium matrix composite;chem icalvapordeposition
TG146.2
A
1007-2373(2016)03-0031-07
10.14081/j.cnki.hgdxb.2016.03.006
2016-02-28
国家自然科学基金(21406052);河北省自然科学基金(E2015202037)
于洋(1988-),男(汉族),硕士生.通讯作者:李海鹏(1977-),男(汉族),副教授.
猜你喜欢
实用手外科杂志(2022年2期)2022-08-31
城市道桥与防洪(2022年3期)2022-05-08
山东陶瓷(2021年5期)2022-01-17
陶瓷学报(2021年5期)2021-11-22
陶瓷学报(2021年1期)2021-04-13
云南化工(2020年11期)2021-01-14
应用化工(2020年9期)2020-09-29
天然产物研究与开发(2018年5期)2018-06-13
中成药(2018年1期)2018-02-02
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
