
不同年龄麦洼牦牛肠道菌群的分离鉴定及其群落结构的变化
2016-02-07 02:50聂远洋刘戎梅官久强罗晓林
中国测试 2016年12期
聂远洋,邓 岳,刘戎梅,官久强,罗晓林,孙 群
(1.四川大学生命科学学院生物资源与生态环境教育部重点实验室,四川 成都 610064;2.泸州职业技术学院,四川 泸州 646000;3.四川省草原科学研究院,四川 成都 611731)
不同年龄麦洼牦牛肠道菌群的分离鉴定及其群落结构的变化
聂远洋1,邓 岳2,刘戎梅1,官久强3,罗晓林3,孙 群1
(1.四川大学生命科学学院生物资源与生态环境教育部重点实验室,四川 成都 610064;2.泸州职业技术学院,四川 泸州 646000;3.四川省草原科学研究院,四川 成都 611731)
为了解麦洼牦牛肠道细菌群落结构组成和功能及其动态变化过程,同时获得具有潜在益生菌活性和应用可能性的肠道菌株,采用不同的培养基在不同的培养条件下分离麦洼牦牛肠道细菌并进行16S rDNA分子鉴定,同时采用变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)技术分析不同年龄麦洼牦牛肠道细菌的群落结构和功能及其差异。结果表明,从不同年龄麦洼牦牛大肠内容物中共分离到90株细菌,其中3株归为放线菌门(Actinobacteria),64株为厚壁菌门(Firmicutes),23株为变形菌门(Proteobacteria);分离到的34株芽孢杆菌属(Bacillus)和14株乳酸杆菌属(Lactobacillus)细菌具有潜在益生菌活性,可作为微生物饲料添加剂补饲麦洼牦牛;对DGGE图谱的UPGMA聚类、PCA和多样性指数分析显示,不同年龄的麦洼牦牛肠道细菌的群落结构有差异,相同年龄的个体相似度较高,而不同年龄的个体相似度较小,较大年龄(2.5和3.5岁)的牦牛肠道菌群多样性大于较小年龄个体(0.5和1.5岁),且分布更均匀,肠道菌群随宿主年龄变化是一个动态变化并趋于稳定的过程;DGGE条带的物种鉴定结果表明,大部分条带为未(能)培养细菌,PCR-DGGE与分离培养法结合可以很好地表征麦洼牦牛肠道菌群的结构和功能。
肠道菌群;牦牛;培养法;16S rDNA;PCR-DGGE
0 引 言
牦牛(Yak,Bos grunniens)是高寒地区的特有牛种,草食性反刍家畜,也是我国的主要牛种之一,主要产于青藏高原海拔3000m以上地区。麦洼牦牛是在川西北高寒生态条件下经长期自然选择和人工选择形成的乳、肉性能良好的草地型牦牛品种,对高寒草地特别是沼泽草地有良好的适应性,是四川省群体数量最大的牦牛地方品种[1]。牦牛具有奶、肉、绒、皮等多种经济用途,且具有风味独特、品质优良的奶和肉,来自高原地区的牦牛产品正以其无污染的特色野味逐渐受到人们的青睐[2]。
牦牛的肠道是其消化和吸收营养物质的主要场所,直接关系到牦牛的营养与健康。其肠道内共生着大量细菌,能进一步分解食物残渣和植物纤维,促进牦牛对营养物质的吸收[3]。肠道正常菌群在一般情况下不会致病,一些肠道细菌还能利用肠道内简单的物质合成维生素B和维生素K等;但由于牦牛的主产区青藏高原海拔较高、气候严寒恶劣、冷季草料匮乏,易导致牦牛掉膘、生病和死亡等冷季损失问题,因此深入了解牦牛肠道微生物的群落结构对牦牛的营养具有十分重要的意义。目前关于系统深入地研究牦牛肠道微生物的群落结构和功能,以及它们的动态变化和与牦牛营养代谢的潜在关联还鲜见报道,少量的研究大都集中于其肠道微生物的分离培养鉴定,且与疾病的防控相关[4-5]。
传统培养法存在只能反映少数微生物的信息、检测结果误差较大、易漏掉具有应用价值的微生物资源等问题[6],但这种方法在分离具有一定特殊功能的目标微生物时非常有用,许多微生物资源的开发与利用还是要依赖传统的微生物培养技术[5,7]。PCRDGGE指纹图谱技术在研究复杂微生物区系的遗传多样性和种群差异等方面具有独特的优势,目前已成为分析微生物群落结构和动态变化广泛采用的技术[8]。而随着大规模测序技术的发展,宏基因组学(Metagenomics)以环境中未培养微生物为研究对象,近年来在微生物生态学和环境微生物学研究中得到了快速的发展[9-10],这一技术在揭示未发现的物种存在,以及鉴定微生物组的部分基因功能和不同微生物的基因功能差异等方面具有更加强大的功能,因此也成为目前肠道微生物研究重点采用的技术[11]。本研究采用不同的培养基在不同的培养条件下分离麦洼牦牛肠道细菌并进行16S rDNA分子鉴定,分析讨论获得的菌株的益生菌活性和应用可能性;同时采用PCR-DGGE技术分析不同年龄麦洼牦牛肠道细菌的群落结构和功能及其差异,分析并讨论肠道菌群随宿主年龄的动态变化过程。
1 材料与方法
1.1 材料
1.1.1 主要试剂
细菌基因组DNA提取试剂盒,北京天根公司;Stool DNA Kit,Gel Extraction Kit,美国OMEGA公司;日本Takara公司;SYBR Green I核酸染料,美国Sigma公司;LB培养基(LB)、脑心浸液肉汤培养基(BHI)和厌氧琼脂培养基(SAA),青岛海博生物技术有限公司。
1.1.2 主要仪器
S1000TMPCR仪,WIDE型电泳槽,PowerPacTMBasic型电泳仪,UniversalHood II型凝胶成像系统,DCodeTMUniversal Mutation Detection System,美国Bio-Rad公司;LEGEND MICRO 21R型高速冷冻离心机,1389型生物安全柜(A2),美国Thermo Fisher Scientific公司;AFZ-1002-U型超纯水仪,艾科浦公司;厌氧密封培养罐(2.5L和7.0L),日本三菱瓦斯化学株式会社;DHP-9162型电热恒温培养箱,上海一恒科学仪器有限公司。
1.2 方法
1.2.1 样品采集
所有麦洼牦牛肠道内容物样品采自四川省阿坝藏族羌族自治州红原县肉联厂,各年龄阶段(0.5,1.5,2.5,3.5岁)的麦洼牦牛均由四川省草原科学研究院规范化养殖,在屠宰时剪取牦牛的大肠(含内容物)并分别冷藏保存,用于分离微需氧菌和完全厌氧菌的样品则迅速放入厌氧密封罐(分别含微氧产气袋和完全厌氧产气袋)中冷藏保存。分别取到各年龄阶段的麦洼牦牛肠道内容物样品各5份。
1.2.2 菌株分离纯化
取麦洼牦牛肠道内容物用无菌水分散后涂布于新鲜制备的LB培养基平板和脑心浸液肉汤培养基平板上,置于37℃恒温培养箱中培养1~2d,待长出单菌落后用接种环挑取单菌落划线转接至新鲜的LB培养基平板和脑心浸液肉汤培养基平板上,按前述方法培养1~2d,如此反复挑取单菌落纯化至得到纯培养,并分别甘油保藏和真空冷冻干燥保藏菌种。
取用于分离微需氧菌和完全厌氧菌的样品用无菌水分散后分别涂布于新鲜制备的厌氧琼脂培养基平板上,迅速置于厌氧培养盒中,向盒子的水槽中倒入少量无菌水,然后分别放入微氧产气包(5%氧气残留)和完全厌氧产气包(无氧气残留),迅速盖上盒子的密封盖,将厌氧盒置于37℃恒温培养箱中培养5~7d,待长出单菌落后用接种环挑取单菌落划线转接至新鲜的厌氧琼脂培养基平板上,按前述方法培养5~7d,如此反复挑取单菌落纯化至得到纯培养,并分别甘油保藏和真空冷冻干燥保藏菌种。
1.2.3 基因组DNA提取
采用TIANGEN公司细菌基因组DNA提取试剂盒提取分离纯化的细菌基因组DNA,采用OMEGA公司Stool DNA Kit试剂盒提取麦洼牦牛肠道内容物样品的总基因组DNA,用1.5%琼脂糖凝胶电泳检测提取结果。
1.2.4 16S rDNA扩增和胶回收
PCR引物序列[8]为:27F 5′-AGAGTTTGATCMTG GCTCAG-3′,1492R 5′-TACGGYTACCTTGTTACGA CTT-3′。50 μL体系:4 μL模板DNA,上下游引物各2μL,25μL 2×Taq PCR MasterMix(Loading dye mix),17 μL ddH2O。扩增条件:94℃预变性5 min;94℃,30 s,58℃,45s,72℃,90s,30个循环;最后72℃,延伸7min。阴性对照用ddH2O作为模板。扩增结束后,用1.5%琼脂糖凝胶电泳检测,以凝胶成像系统采集图片并进行结果分析。采用OMEGA公司的Gel Extraction Kit回收琼脂糖凝胶中的16S rDNA,并用1.5%琼脂糖凝胶电泳检测回收结果。
PCR引物序列[8]为:HDA1-GC 5′-CGCCCGGGG CGCGCCCCGGGCGGGGCGGGGGCACGGGGGGACT CCTACGGGAGGCAGCAGT-3′,HDA2 5′-GTATTAC CGCGGCTGCTGGCAC-3′。50 μL体系:4 μL模板DNA(胶回收后的肠道内容物总基因组DNA的16S rDNA扩增产物),上下游引物各2 μL,25 μL 2×Taq PCR MasterMix(Loading dye mix),17μL ddH2O。扩增条件:94℃预变性5min;94℃,30s,53℃,30s,68℃,30s,30个循环;最后68℃,延伸7min。阴性对照用ddH2O作为模板。扩增结束后,用1.5%琼脂糖凝胶电泳检测,以凝胶成像系统采集图片并进行结果分析。
1.2.6 DGGE
制备8%聚丙烯酰胺凝胶,变性梯度浓度为35%~65%,呈线性增加,梯度方向与电泳方向平行,加入20μL含有适量上样缓冲液的16S rDNA V3区PCR扩增产物。电泳时,温度为60℃,电压为120V,运行6h。电泳后用SYBR Green I进行染色,染料用1×TAE以1∶10 000稀释,每次染液用量15 mL,染色15min,共染3次。染色完后用凝胶成像系统采集图片并用Quantity One 4.6.9软件分析结果。
1.2.7 物种鉴定
将胶回收后的各分离菌株的16S rDNA送至英潍捷基(上海)贸易有限公司测序,获得的序列分别在NCBI BLAST上比对并分析结果。将DGGE凝胶上的特征条带切下,加入50~100 μL ddH2O,在4℃冰箱放置过夜,使胶条中的DNA释放出来,以此为模板,用不含GC夹子的V3区引物扩增,反应体系和条件同前1.2.5,扩增产物送英潍捷基(上海)贸易有限公司测序,将获得的序列在NCBI BLAST数据库中比对后获得物种信息。
1.2.8 统计分析
采用Quantity One 4.6.9软件将DGGE图谱的DNA条带亮度数字化,根据香农指数(H′)、丰富度(S)和均匀度(EH)的计算公式分别计算得到麦洼牦牛肠道菌群的香农指数、丰富度和均匀度,以分析各样品中细菌的种群多样性及分布的均匀性。各指数的计算式如下式所示:
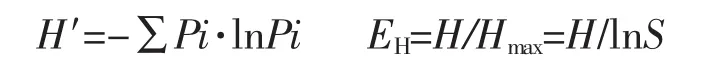
式中:Pi——某一条带的亮度与同泳道中所有条带总亮度的比值;
整个一体化五防系统内所有五防主/子站、电脑钥匙均具备黑匣子功能,即:系统对全时段所有倒闸操作情况进行记录,包括各操作任务的已/未执行项及时间、异常操作等详细信息;并可按操作类型、电压等级、操作时间等任意定制关键字对记录数据进行查寻、调阅;记录在电脑钥匙中的信息可调阅但无法删除;最大记录项数2000项,掉电记忆达5年。系统提供将所需数据根据可调模板制作电子数据并打印。
S——每一泳道中总的条带数目。
实验数据均以均值±标准差表示,采用SPSS 17.0软件的One-way ANOVA进行方差分析,并用LSD法和Dunnett’s T3法进行事后两两比较分析。采用Canoco for Windows 4.5软件进行PCA分析。
2 结果与分析
2.1 麦洼牦牛肠道细菌的分离鉴定结果
采用LB和BHI培养基分离需氧菌,采用SAA培养基分别在微氧(5%氧气)和无氧条件下分离微需氧菌和厌氧菌,分离菌株的鉴定结果和分类见表1。从不同年龄麦洼牦牛大肠内容物中共分离到90株细菌,主要归为放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)和变形菌门(Proteobacteria),其中绝大多数菌株属于厚壁菌门(64株)和变形菌门(23株)细菌,放线菌门细菌仅有3株,而拟杆菌门(Bac teroidetes)细菌则未分离到。在属的水平上,85%以上的菌株归属于芽孢杆菌属(Bacillus)、埃希氏杆菌属(Escherichia)、乳杆菌属(Lactobacillus)和链球菌属(Streptococcus)这4个属,分别有34,20,14,10株细菌。从分离培养条件来看,得到的需氧菌数量(57株)要远高于微需氧菌(12株)和厌氧菌(21株)。
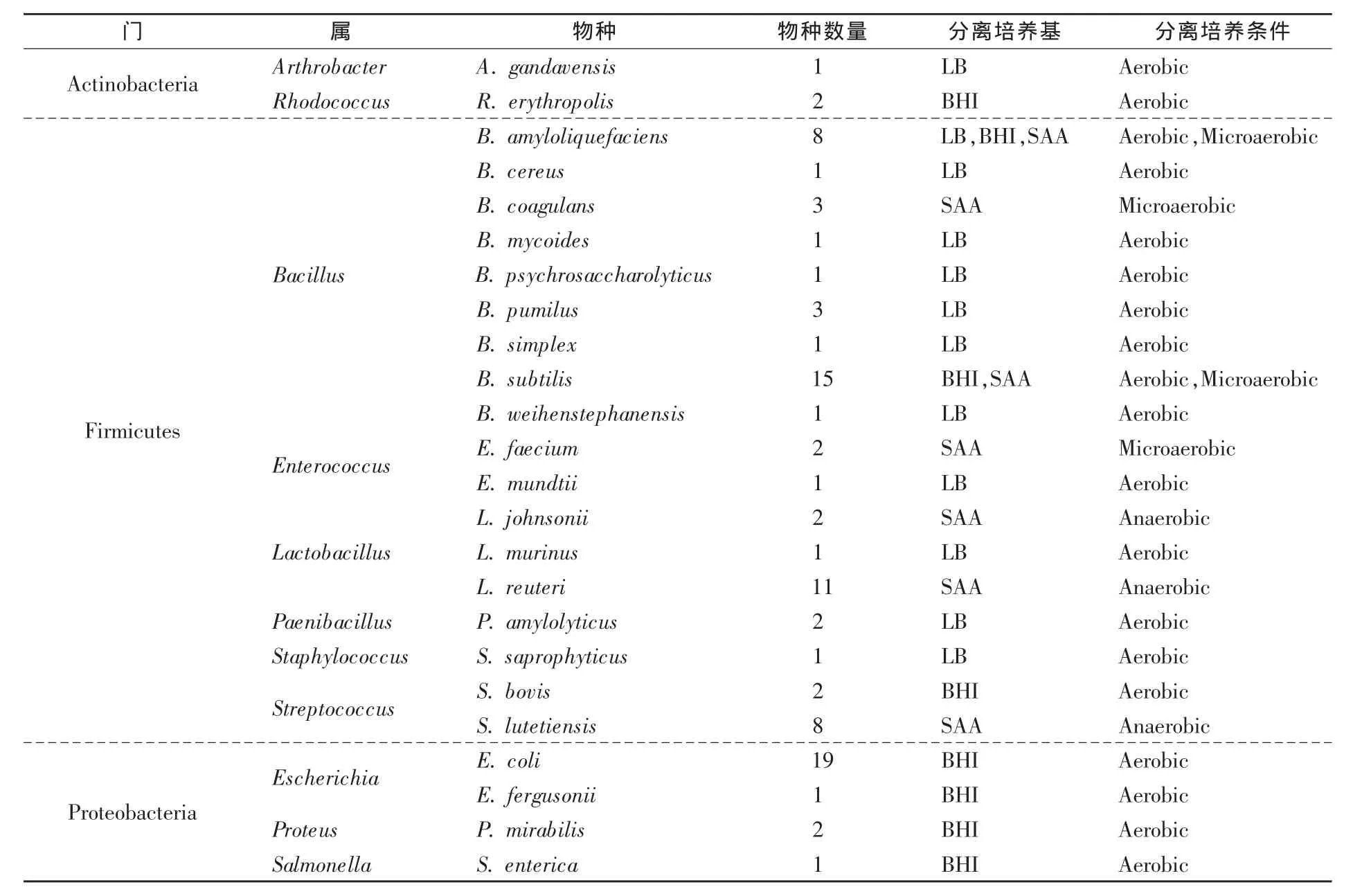
表1 基于16S rRNA基因序列比对后的麦洼牦牛肠道菌株亲缘关系分类1)
2.2 麦洼牦牛肠道菌群16S rDNA V3区DGGE图谱
不同年龄(0.5,1.5,2.5,3.5岁)麦洼牦牛肠道菌群的DGGE图谱如图1所示,图中各泳道的条带数量和亮度的差异反映了各样品中肠道细菌种群多样性和含量的差异。总体来看,在条带数量上2.5和3.5岁麦洼牦牛最多,其次是1.5岁,而0.5岁最少,表明4个不同年龄阶段的麦洼牦牛其肠道细菌种群多样性存在一定的差异;在特征优势条带上0.5和1.5岁麦洼牦牛明显多于2.5岁,也就是说0.5和1.5岁麦洼牦牛含有更多丰度较高的优势细菌种群,而2.5和3.5岁麦洼牦牛肠道菌群的分布较为均一,表明麦洼牦牛在生长过程中肠道菌群的多样性及分布趋于稳定和均一。
2.3 UPGMA聚类分析和PCA分析
采用Quantity One 4.6.9软件的UPGMA算法对DGGE图谱进行聚类分析,以了解不同年龄麦洼牦牛肠道细菌物种类型和丰度在总体水平上相似度的差异,聚类结果如图2(a)所示。从图中可以看出,同一年龄的麦洼牦牛在肠道细菌物种类型和丰度的总体水平上最先聚为一类,且相似值达到0.70以上,说明细菌群落结构差异较小,而不同年龄麦洼牦牛的聚类距离最远,细菌群落结构差异较大。采用Quantity One 4.6.9软件将DGGE图谱的每一个泳道(样品)的每一个条带的亮度转换为数值,并用Canoco for Windows 4.5软件进行PCA分析,结果如图2(b)所示。从图中可以看出,同组样品间距离较小,能大致聚在一起,而不同组间则距离较远,0.5和1.5岁两组间的个体样本距离较近,而与2.5和3.5岁组内个体样本距离都较远,与DGGE图谱和UPGMA聚类分析结果一致。
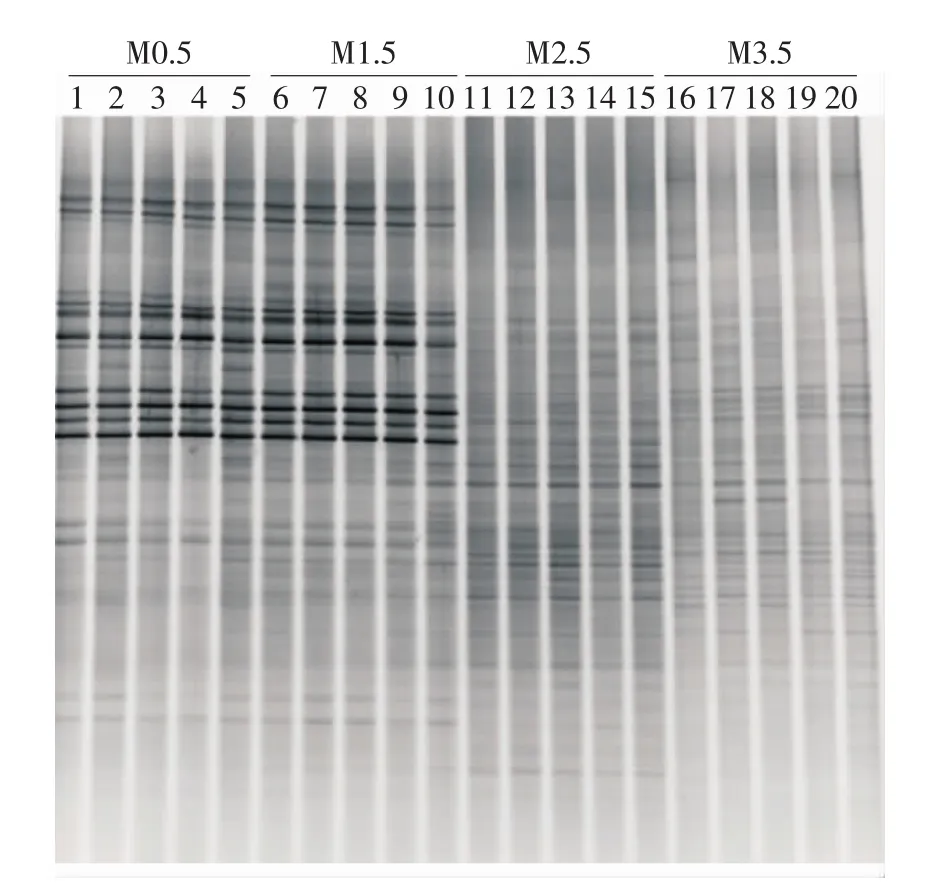
图1 麦洼牦牛肠道菌群16S rDNA V3区DGGE图谱

图2 不同年龄麦洼牦牛肠道菌群DGGE图谱的UPGMA聚类分析和PCA分析
2.4 不同年龄麦洼牦牛肠道菌群的多样性指数分析
分别计算得到不同年龄麦洼牦牛肠道菌群的香农指数(H′)、丰富度(S)和均匀度(EH),以分析各样品中细菌种群多样性及分布的均匀性,结果如表2所示。从表中可以看出3.5和2.5岁麦洼牦牛肠道菌群的香农指数、丰富度和均匀度均显著性高于1.5和0.5岁(P<0.05),表明不同年龄阶段的麦洼牦牛肠道细菌种群多样性和分布均一性存在显著性差异,与直观的DGGE图谱分析结果一致;而0.5和1.5岁麦洼牦牛肠道菌群的香农指数、丰富度和均匀度则没有显著性差异(P>0.05);2.5和3.5岁麦洼牦牛肠道菌群的香农指数、丰富度和均匀度也没有显著性差异(P>0.05)。

表2 不同年龄麦洼牦牛肠道菌群的香农指数、丰富度和均匀度显著性差异分析(N=5)1)
2.5 麦洼牦牛肠道菌群DGGE图谱的特征条带回收及物种鉴定
从不同年龄麦洼牦牛肠道菌群DGGE图谱中切胶回收了42个特征条带,通过PCR扩增、胶回收、测序和NCBI BLAST序列比对后得到各条带代表的物种信息,如表3所示。麦洼牦牛肠道菌群绝大部分为未(能)培养细菌,其中相对丰度较多的主要有梭菌属(Clostridium)、假单胞菌属(Pseudomonas)、消化链球菌属(Peptostreptococcus)和瘤胃球菌科(Ruminococcaceae)的细菌,这些细菌也主要存在于幼年时期(0.5和1.5岁)麦洼牦牛的肠道中,它们构成了幼年麦洼牦牛的肠道优势菌群。而随着麦洼牦牛的成长(2.5和3.5岁),这些优势菌群的丰度逐渐减少,另外一些新的菌属的细菌被检测到,如微杆菌属(Microbacterium)、疣微菌属(Verrucomicrobia)、放线菌属(Actinobacterium)、Akkermansia sp.、脱硫弧菌属(Desulfovibrio)、葡萄球菌属(Staphylococcus)、毛螺菌科(Lachnospiraceae)、优杆菌科(Eubacteriaceae)、绿弯菌纲(Chloroflexi)等细菌,这些细菌也是麦洼牦牛肠道中的正常菌群,它们增加了其宿主肠道菌群的多样性和均匀性,更有助于成年(>2岁)麦洼牦牛对草料类食物的消化和营养的吸收。

表3 麦洼牦牛肠道菌群DGGE图谱的特征条带序列比对结果
3 讨论与结论
3.1 麦洼牦牛肠道菌群中的潜在益生菌资源
本研究中采用不同培养基和不同的培养条件从麦洼牦牛大肠内容物中分离可培养细菌,其中分离到的最多的是芽孢杆菌属(Bacillus)的细菌,共有34株,常见的有枯草芽孢杆菌、蜡样芽胞杆菌、解淀粉芽孢杆菌、甲基营养型芽孢杆菌、韦氏芽孢杆菌、简单芽孢杆菌、短小芽孢杆菌和凝结芽孢杆菌等。我国于1994年法定乳酸杆菌、芽孢杆菌、粪链球菌、酵母菌和双歧杆菌等是可用于微生态制剂生产的主要菌种[12]。因为芽孢杆菌的芽孢可耐长期贮存,所以一般将芽孢作为益生菌产品开发生产,常用于饲料添加剂的芽孢杆菌属的细菌主要有枯草芽孢杆菌、蜡状芽孢杆菌和凝结芽孢杆菌等[13]。乳酸杆菌是其作为微生物饲料添加剂而研究最多且应用最广泛的一类益生菌[14],其在动物肠道内繁殖属于正常菌种,而且可产生多种抑制性化合物和对抗性物质以及有机酸等,这些有机酸本身就是动物生长所需的营养物质,同时还能降低肠道内的pH值,从而抑制其他致病菌和腐败菌的增殖。因此,本研究中分离到的大量芽孢杆菌属和乳酸杆菌属的细菌可在后续的实验中检测其益生菌活性和安全性,并作为益生菌剂直接补饲麦洼牦牛或者添加到青贮饲草中再补饲麦洼牦牛,以提高其生产性能和减少在冷季的损失。
当然,培养法中所使用的分离培养基的种类及其营养成分的丰富程度也对分离到的细菌种类和数量有直接的影响,比如本研究中所使用的脑心浸液肉汤培养基,其营养成分相对丰富,因此相对另外两种培养基而言,其分离到的菌群种类和数量都最多。但是动物肠道的可培养微生物毕竟只占很小的一部分,要了解麦洼牦牛整个肠道微生物的群落结构和功能,还需要其他分子生物学的非培养法的结合研究,而DGGE条带的鉴定结果表明大部分的条带为未(能)培养细菌,因此两种方法结合可更加全面地了解麦洼牦牛肠道菌群的结构和功能。
3.2 不同年龄麦洼牦牛肠道菌群的群落结构动态变化
对DGGE图谱中不同样本的UPGMA聚类、PCA和多样性指数分析,结果表明不同年龄的麦洼牦牛其肠道菌群的群落结构都有一定的差异,表现为相同年龄的个体相似度较高,聚类和PCA结果也聚为一类,而不同年龄间的个体相似度较小,聚类和PCA距离较远;在多样性指数(香农指数、丰富度和均匀度)的差异上,0.5和1.5岁之间无显著性差异,2.5和3.5岁之间也无显著性差异,而较大年龄组(2.5和3.5岁)显著性高于较小年龄组(0.5和1.5岁),表明较大年龄的麦洼牦牛其肠道菌群的多样性大于较小年龄个体,且分布更均匀,肠道菌群随宿主的年龄变化是一个动态变化并趋于稳定的过程。分析其原因可能是食物的影响,动物从出生到成年其肠道菌群是一个从不断定植到逐渐稳定的过程[15],麦洼牦牛在较小年龄时未完全断奶,奶和草同时进食,而成年后(一般2岁左右)只食草料,肠道菌群也已完成定植,因此表现为更高的多样性和均匀性。
4 结束语
肠道菌群可以受到来自宿主方面的很多影响,如食物、年龄、抗生素的使用情况、健康状况等,同时也可以受很多环境因子的影响,如生活地域、季节和饲养条件等[8]。本研究采用PCR-DGGE技术分析了不同年龄麦洼牦牛肠道细菌的群落结构及其差异,证实了麦洼牦牛肠道菌群随宿主的年龄变化是一个动态变化并趋于稳定的过程,表现为成年麦洼牦牛的肠道菌群具有更高的多样性和均匀性。并采用不同的培养基在不同的培养条件下分离到90株麦洼牦牛肠道细菌,其中34株芽孢杆菌属和14株乳酸杆菌属细菌具有潜在益生菌活性,经验证后可作为微生物饲料添加剂用于补饲麦洼牦牛,以提高其生产性能和减少在冷季的损失。
[1]黄彩霞,高媛,孙宝忠,等.牦牛品种品质研究进展[J].肉类研究,2012,26(9):30-34.
[2]徐培伦,张正萍,彭宇鸿,等.麦洼牦牛肉品质相关特性研究[J].四川动物,2015,34(5):759-763.
[3]左愈臻,高世杰,邵建华,等.成年牦牛小肠结构及黏膜免疫相关细胞数量变化研究[J].畜牧兽医学报,2011,42(12):1776-1781.
[4]任丽倩,于学辉,宋定州,等.牦牛大肠杆菌的分离鉴定及系统发育分析[J].中国畜牧兽医,2012,39(1):168-172.
[5]董映辉,张朝辉,毛全富,等.甘孜州牦牛沙门氏菌的分离和鉴定[J].四川畜牧兽医,2011,244(2):27-29.
[6]BLAUT M. Ecology and physiology of the intestinal tract[M].Berlin:Springer,2011:247-272.
[7]张贝贝,赵晓兵,何丽丽,等.牛瘤胃细菌产木聚糖酶菌株的筛选及酶学性质研究[J].河南农业科学,2014,43(4):128-132.
[8]LI M J,ZHOU M,ADAMOWICZ E,et al.Characterization of bovine ruminal epithelial bacterial communities using 16S rRNA sequencing,PCR-DGGE,and qRT-PCR analysis[J].Veterinary Microbiology,2012,155(1):72-80.
[9]刘海燕,常玉梅.宏基因组学及在人体微生物研究上的应用[J].中国现代医学杂志,2012,22(8):51-54.
[10]MACKELPRANG R,WALDROP M P,DEANGELIS K M,et al.Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw[J].Nature,2011,480(7377):368-371.
[11]QIN J J,LI Y R,CAI Z M,et al.A metagenomewide association study of gut microbiota in type 2 diabetes[J].Nature,2012,490(7418):55-60.
[12]徐鹏,董晓芳,佟建明.微生物饲料添加剂的主要功能及其研究进展[J].动物营养学报,2012,24(8):1397-1403.
[13]刁其玉.微生物制剂在幼龄反刍动物营养与饲料中的应用[J].动物营养学报,2014,26(10):3159-3167.
[14]丁轲,余祖华,程相朝,等.高产纤维素酶益生乳酸杆菌的分离鉴定[J].饲料与畜牧:新饲料,2010(11):37-40.
[15]ZHAO W,WANG Y,LIU S,et al.The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments[J].Plos One,2015,10(2):117-441.
(编辑:莫婕)
Isolation and identification of gut microbiota with community structure changes in different ages of Maiwa yaks
NIE Yuanyang1,DENG Yue2,LIU Rongmei1,GUAN Jiuqiang3,LUO Xiaolin3,SUN Qun1
(1.Key Laboratory of Bio-resource and Bio-environment,College of Life Sciences,Sichuan University,Chengdu 610064,China;2.Luzhou Vocational and Technical College,Luzhou 646000,China;3.Sichuan Grassland Science Academy,Chengdu 611731,China)
To understand the community structure composition,functions and dynamic changes of Maiwa yaks’gut microbiota and obtain the gut bacterial strain of Maiwa yaks with Potential probiotic activity and application possibility,different culture mediums were used to separate the intestinal bacteria of Maiwa yaks under different culture conditions,and 16S rDNA molecular identification was also carried out.In the meantime,denaturing gradient gel electrophoresis(DGGE)technology was also applied to analyze the community structure and function and the difference of intestinal bacteria of Maiwa yaks at different ages.The results showed that totally 90 strains of bacteria were separated from the gut content of Maiwa yaks at different ages,in which 3,64,and 23 strains were classified as the phylum ofActinobacteria, Firmicutes and Proteobacteria respectively.Totally 34 strains of the genus Bacillus and 14 strains of the genus Lactobacillusbacteria with potential probiotic activity were also separated,and they can be used as microbial feed additives to Maiwa yak supplementary feeding.Analysis of UPGMA clustering,PCA,and diversity index of DGGE spectrum indicated that the community structure of gut microbiota at different ages is different,and the similarity between animals of the same ages is high,while the similarity between different ages of animals is low.The diversity of gut microbiota of elder groups of Maiwa yaks(2.5 and 3.5 years)is significantly higher than that of younger groups(0.5 and 1.5 years)but with more homogeneous distribution,which indicates that the change of gut microbiota along with the age change of host is a dynamic process tending to be stable.Species identification results of DGGE bands showed that most of the bands were uncultured bacteria,further indicating that the method of combining PCR-DGGE with plate-culture method can be a more comprehensive way to characterize the structure and function of gut microbiota of Maiwa yaks.
gut microbiota;yak;culture-dependent method;16S rDNA;PCR-DGGE
A
:1674-5124(2016)12-0053-07
10.11857/j.issn.1674-5124.2016.12.012
2016-08-09;
:2016-09-21
四川省科技支撑计划课题(2016NZ0005)
聂远洋(1986-),男,重庆市人,博士,研究方向为资源微生物。
孙 群(1967-),女,四川成都市人,教授,博士,研究方向为微生物技术与食品安全。
猜你喜欢
中老年保健(2022年2期)2022-08-24
青海湖(2022年3期)2022-06-09
滇池(2022年5期)2022-04-30
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年3期)2022-04-19
散文诗(2021年22期)2022-01-12
湖南饲料(2021年4期)2021-10-13
小哥白尼(野生动物)(2020年9期)2021-01-18
科学(2020年4期)2020-11-26
农药科学与管理(2019年6期)2019-11-23
