
吡哆醛催化酮酸转氨化的研究
2016-02-05 05:06:00赵濬宇赵宝国
上海师范大学学报·自然科学版 2016年6期
陈 静, 赵濬宇, 赵宝国
(上海师范大学 生命与环境科学学院,上海 200234)
吡哆醛催化酮酸转氨化的研究
陈 静, 赵濬宇, 赵宝国
(上海师范大学 生命与环境科学学院,上海 200234)
吡哆醛及其衍生物是酶催化转氨化的活性中心.在酶催化的转氨化过程中涉及到碱去质子化和酸质子化的过程,而酸或(和)碱的引入有可能降低转氨化反应的活化能.因此通过在吡哆醛上引入胺、酸或脲等基团可能得到更加有效的转氨化催化剂.设计合成了一系列的带有不同官能团的吡哆醛催化剂.这些吡哆醛在α-酮酸转氨化反应中展现出了较高的活性且能以较好的收率合成多种氨基酸.该反应条件温和,易于操作,是一种合成α-氨基酸的好方法.
酮酸; 转氨化; α-氨基酸; 吡哆醛
0 引 言
氨基酸是生物功能大分子蛋白质的基本组成单元,是构成动物营养所需蛋白质的基本物质.自20世纪50年代开始,氨基酸的应用范围不断扩大,形成了一个朝气蓬勃的新兴工业体系,被称为氨基酸工业,生产技术日新月异,品种和产量逐年增加,1969年世界氨基酸的总产量约为25万吨,1979年为40万吨,10年内增加约1.7倍,至1989年调查统计总产出55万吨[1].
在生物体内,酶催化的转氨化是合成氨基酸的重要途径,该反应是通过转氨酶催化实现的[2-4].转氨酶的活化中心是维生素B6,包括吡哆醛和吡哆胺及其磷酸酯衍生物.转氨化反应实现了由α-酮酸到α-氨基酸的转化.酶催化酮酸的转氨化具有反应速度快,对映选择性高,底物专一性强等优点.但是大多数转氨酶不易得到,价格昂贵,另外由于酶专一性过强的特点又限制了底物适用范围,这些不足限制了酶催化酮酸转氨化的应用.所以发展维生素B6及其衍生物催化的生物模拟转氨化反应具有重要意义.该研究很早就被化学家们重视[5-60],如1952年,Snell等[10-11]发现吡哆醛1与一系列的氨基酸2之间可以发生转氨化,生成相应的吡哆胺3和α-酮酸4(图1).
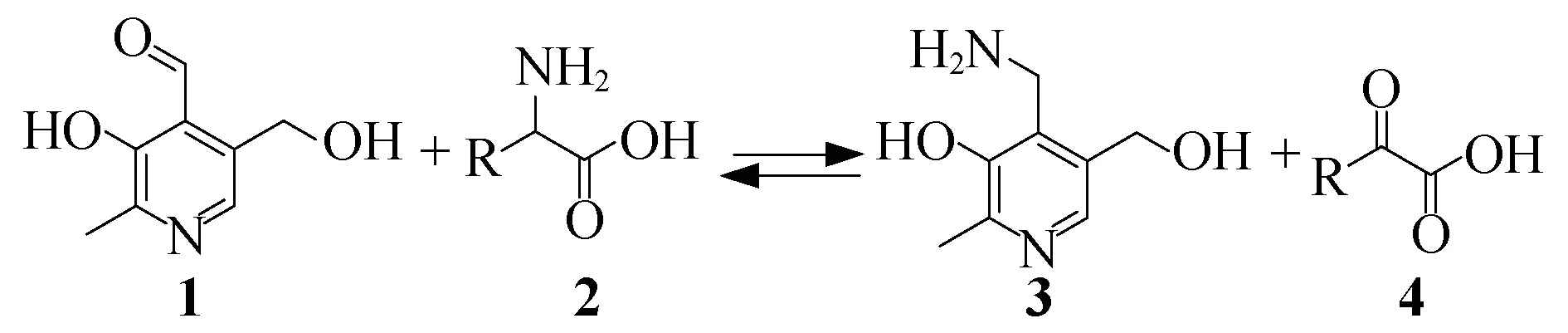
图1 Snell等[10-11]对转氨化反应的初步探索
从此拉开了生物模拟酮酸转氨化反应研究的序幕[10-60].生物模拟酮酸转氨化已经进行了大量的研究,但是到目前为止,生物模拟转氨化,尤其是以维生素B6的衍生物为催化剂模拟转氨化远没有发展成熟,具有较大的发展空间和应用潜力,需要进一步的探索.
在酶催化的转氨化过程中,涉及到碱去质子化和酸质子化的过程,因此,设想在吡哆醛分子上合适的位置引入适当的碱或(和)酸的基团,在转氨化反应中参与去质子或(和)质子化的过程,有可能降低反应的活化能,提高催化活性和选择性.正是基于这一思路,本文作者通过在吡哆醛的侧链上的不同位置引入不同官能团,设计并合成了一系列的吡哆醛催化剂,并考察了它们在α-酮酸转氨化反应中的催化性能.
1 实验部分
1.1 实验通则
特别注明除外,溶剂都以标准方法处理.1H NMR和13C NMR在400 MHz 和600MHz Bruker核磁共振仪上测定.甲苯、四氢呋喃、苯等经钠丝干燥处理后使用;二氯甲烷、N,N-二甲基甲酰胺等经CaH2干燥处理后使用;CH3CN经五氧化二磷干燥处理后使用;三乙胺 (TEA) 经氢氧化钾干燥处理后使用.固体商业试剂除非特别说明外,均未经纯化直接使用;液体试剂均经蒸馏纯化后使用.
1.2 合成方法
1.2.1 催化剂9的合成
催化剂9合成路线如图2所示,以商业化的吡哆素盐酸盐1为起始原料,经过五步反应,分别合成了7种催化剂9a-g.首先化合物1在硫酸的作用下与丙酮反应,得到丙酮叉保护的吡哆素5.化合物5在二异丙基乙基胺作碱的条件下,和甲磺酰氯反应生成具有较好离去性能的甲磺酸酯6.6与不同的硫醇化合物进行反应得到化合物7,该化合物在乙酸和水中110 ℃的条件下反应4 h脱去丙酮叉保护基,再用二氧化锰氧化即可得到吡哆醛催化剂9.催化剂9a中引入长链,目的是考察脂溶性官能团对转氨化反应催化活性的影响;催化剂9b-f中分别引入了带有酸性质子的官能团,目的是考察不同酸度的质子对催化活性的影响;催化剂9g中引入了NMe2,目的是考察碱性基团对催化活性的影响.
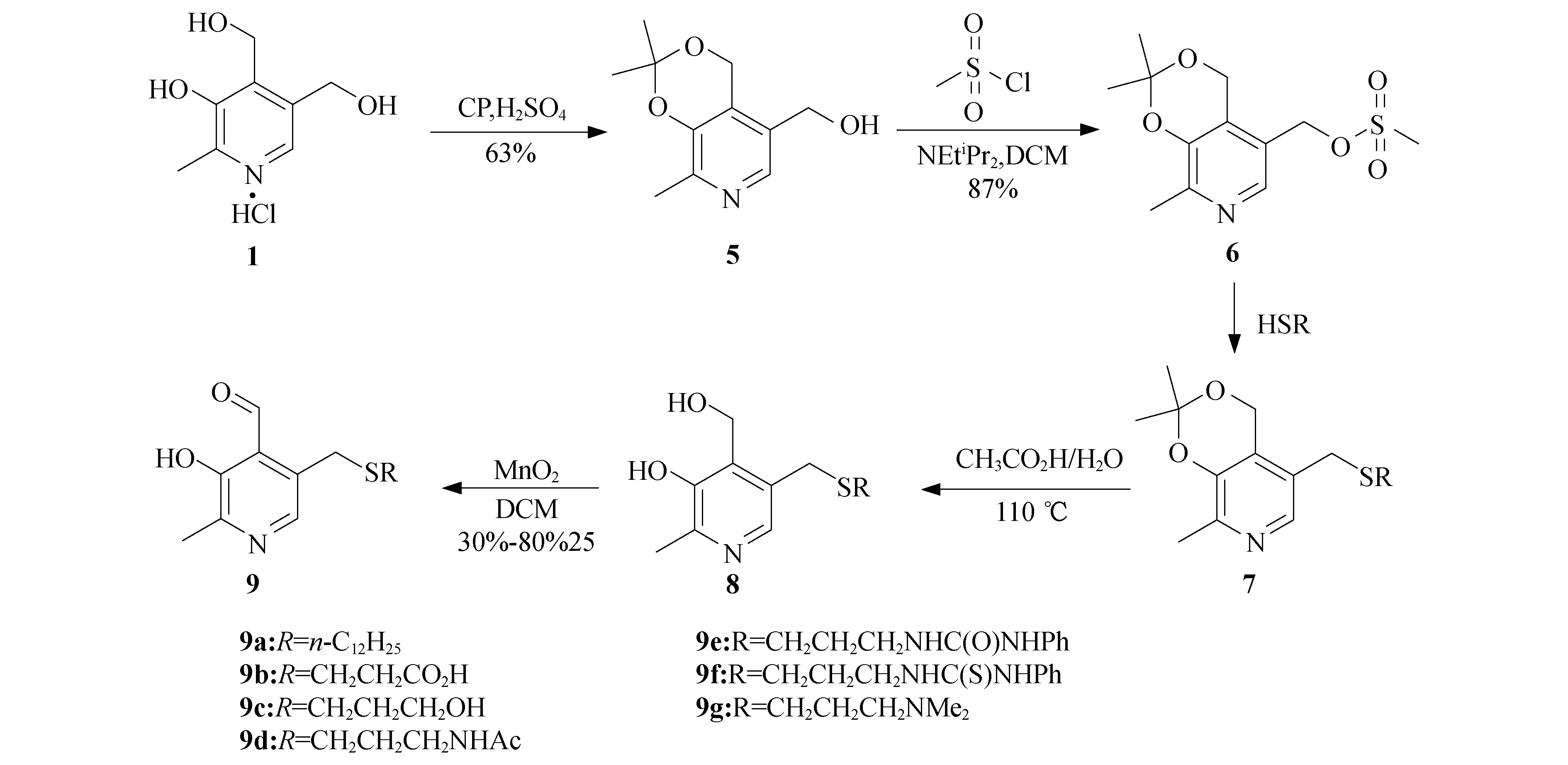
图2 催化剂9a-g的合成
9a:黄色固体;收率80%;1H NMR (400 MHz,CDCl3) δ 11.48 (s,1H),10.52 (s,1H),7.93 (s,1H),3.90 (s,2H),2.50 (s,3H),2.45 (t,J=5.2 Hz,2H),1.59-1.52 (m,2H),1.36-1.20 (m,18H),0.87 (t,J=4.8 Hz,3H);13C NMR (100 MHz,CDCl3) 195.9,154.4,151.7,139.9,131.1,119.6,31.97,31.96,29.69,29.68,29.63,29.54,29.51,29.4,29.2,28.8,22.7,18.8,14.2.
9b:黄色固体;收率30%;1H NMR (400 MHz,DMSO) δ 12.31 (brs,10.51 (s,1H),8.04 (s,1H),4.11 (s,2H),2.64 (t,J=7.6 Hz,2H),2.53 (t,J=7.6 Hz,2H),2.44 (s,1H);13C NMR (100 MHz,CDCl3) δ 196.4,172.8,153.4,150.3,139.7,131.0,119.8,34.0,28.6,26.3,18.2.
9c:黄色固体;收率43%;1H NMR (300 MHz,CDCl3) δ 11.48 (s,1H),10.51 (s,1H),7.93 (s,1H),3.92 (s,2H),3.71 (t,J=6.0 Hz,2H),2.59 (t,J=6.9 Hz,2H),2.50 (s,3H),1.83 (tt,J=6.9,6.0 Hz,2H);13C NMR (100 MHz,CDCl3) δ 196.6,153.1,149.8,140.1,132.2,121.2,59.5,32.2,28.5,27.8,18.7.
9d:黄色固体;收率43%;1H NMR (400 MHz,CDCl3) δ 11.48 (s,1H),10.50 (s,1H),7.91 (s,1H),5.78 (brs.1H),3.91 (s,2H),3.35-3.26 (m,2H),2.50 (s,3H),2.48 (t,J=7.2 Hz,2H),1.94 (s,3H),1.83-1.73 (m,2H);13C NMR (100 MHz,CDCl3) δ 195.8,170.5,154.2,151.6,139.6,130.7,119.5,38.4,29.3,29.1,28.8,23.1,18.6.
9e:黄色固体,收率37%;1H NMR (400 MHz,CDCl3) δ 11.48 (brs,1H),10.46 (s,1H),7.90 (s,1H),7.48 (s,1H) 7.32-7.19 (m,4H),7.06-6.96 (m,1H),5.50 (t,J=5.2 Hz,1H),3.84 (s,2H),3.29-3.20 (m,2H),2.49 (s,3H),2.44 (t,J=7.2 Hz,2H),1.78-1.66 (m,2H);13C NMR (100 MHz,CDCl3) 195.9,156.7,154.5,151.8,139.6,139.0,131.0,129.2,123.3,120.4,119.7,39.0,29.7,29.3,29.2,18.7.
9f:黄色固体;收率22%;1H NMR (400 MHz,CDCl3) δ 11.33,(brs,1H),10.39 (s,1H),7.85 (s,1H),7.36-7.24 (m,2H),7.18-7.10(m,3H),6.46 (s,1H),3.85 (s,2H),3.65-3.55 (m,2H),2.48-2.36 (m,5H),1.85-1.76 (m,2H);13C NMR (100 MHz,CDCl3) δ 195.9,156.7,154.5,151.8,139.6,139.0,131.0,129.2,123.3,120.4,119.7,39.0,29.7,29.3,29.2,18.7.
9g:黄色固体;收率51%;1H NMR (400 MHz,CDCl3) δ 10.49 (s,1H),7.92 (s,1H),3.91 (s,2H),2.58-2.40 (m,7H),2.28 (s,6H),1.84-1.72 (m,2H).
1.2.2 催化剂12的合成
按照图3所示的方法,发展了催化剂12.化合物5与苯酚在偶氮二甲酸二乙酯(DEAD)和三苯基磷存在下发生缩合,以很好的产率得到化合物10.化合物10在乙酸和水中110 ℃反应4 h,脱去丙酮叉保护基得到化合物11,再经过二氧化锰氧化以较好的产率得到吡哆醛催化剂12.
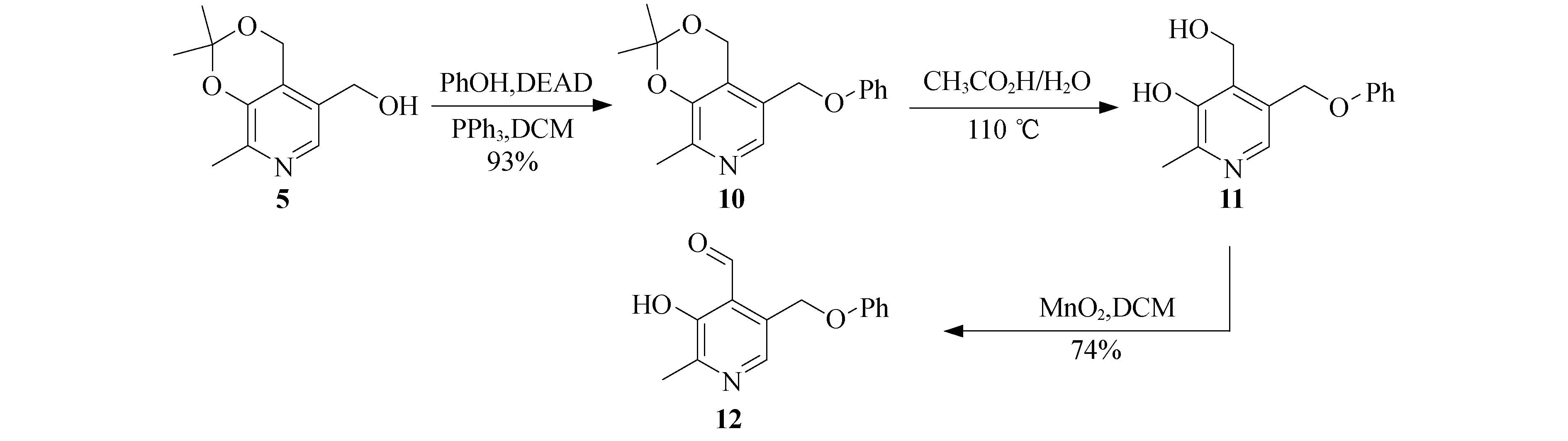
图3 催化剂12的合成
12:黄色固体;收率75%;1H NMR (600 MHz,CDCl3) δ 11.45 (s,1H),10.34 (s,1H),8.12 (s,1H),7.26(dd,J=7.8,7.2 Hz,2H),6.96 (t,J=7.2 Hz,1H),6.92 (d,J=7.8 Hz,2H),5.21 (s,2H),2.50 (m,3H);13C NMR (100 MHz,CDCl3) δ 196.8,157.8,154.3,153.3,139.6,129.9,128.7,122.1,120.6,114.9,65.0,22.1.
1.2.3 α-酮酸的转氨化(以α-酮酸4a的转氨化为例)
取5 mL的反应瓶,向瓶中称取酮酸4a(45.6 mg,0.20 mmol),催化剂12 (2.4 mg,0.010 mmol),2,2-二苯基氨基酸13a(47.7 mg,0.21 mmol),再向瓶中加入MeOH(0.90 mL)和水(0.10 mL),加入磁子,塞好瓶塞,置入25 ℃恒温油浴中反应2 d.停止反应,将瓶中反应物转移到25 mL茄型瓶中,加入10 mL甲醇使瓶中固体全部溶解,再加入0.2 g硅胶,置于25 ℃水浴的旋蒸中旋干,干法上柱.柱层析(silica gel,EtOH/ethyl acetate/25%~28% ammonia solution=100∶58∶16)得到产品氨基酸2a(44.1 mg,yield=96%).

图4 α-酮酸4a的转氨化
2a:白色固体;1H NMR (400 MHz,D2O with 2 equiv.of KOH) δ 8.08-7.96 (m,1H),7.88-7.78 (m,1H),7.75-7.65 (m,1H),7.55-7.20 (m,4H),3.30-3.21 (m,1H),3.06-2.92 (m,2H),1.98-1.75 (m,2H).
2b:白色固体;1H NMR (600 MHz,D2O with 2 equiv.of KOH) δ 3.07 (dd,J=6.4,6.0 Hz,1H),1.50-1.35 (m,2H),1.22-1.06 (m,12H),0.72 (t,J=6.4 Hz,3H).
2c:白色固体:1H NMR (400 MHz,D2O) δ 7.36 (dd,J=7.6 Hz,2H),7.30 (d,J=7.6 Hz,2H),7.25 (t,J=7.6 Hz,1H),3.25 (dd,J=6.4,6.0 Hz,1H),2.64 (t,J=8.0 Hz,2H),1.98-1.76 (m,2H).
2d:白色固体;1H NMR (600 MHz,D2O with 2 equiv.of KOH) δ 6.80 (s,2H),3.20 (t,J=9.0 Hz,1H),2.60-2.50 (m,2H),2.21 (s,6H),2.15 (s,3H),1.68-1.55 (m,2H).
2e:白色固体;1H NMR (400 MHz,D2Owith 2 equiv.of KOH) 3.19 (dd,J=8.4,6.0 Hz,1H),1.66-1.53 (m,1H),1.45-1.25 (m,2H),0.84 (t,J=6.8 Hz,6H).
2g:白色固体;1H NMR (600 MHz,D2Owith 2 equiv.of KOH) δ 7.80 (d,J=8.4 Hz,1H),7.79 (d,J=9.0 Hz,1H),7.71 (s,1H),7.46 (d,J=8.4 Hz,1H),7.30 (s,1H),7.17 (d,J=9.0 Hz,1H),3.91 (s,3H),3.05-2.95 (m,2H),2.09-2.02 (m,1H),1.82-1.73 (m,1H),1.30 (d,J=7.2 Hz,3H).
2h:白色固体;1H NMR (600 MHz,D2Owith 2 equiv.of KOH) δ 7.40-7.25 (m,8H),7.23-7.14 (m,2H),4.08 (t,J=12.0,1H),2.99 (dd,J=12.0,9.0 Hz,1H),2.48-2.36 (m,1H),2.18-2.06 (m,1H).
2i:白色固体;1HNMR (400 MHz,D2Owith 2 equiv.of KOH) δ 7.29-7.14 (m,5H),3.68 (dd,J=9.2,4.4 Hz,1H),3.23 (dd,J=10.4,4.4 Hz,1H),2.90 (dd,J=10.4,9.2 Hz,1H).
2k:白色固体;1H NMR (400 MHz,D2Owith 2 equiv.of KOH) δ 3.27 (dd,J=6.4,6.0 Hz,1H),1.67 (dd,J=14.0,6.4 Hz,1H),1.33 (dd,J=14.0,6.0 Hz,1H),0.88 (s,9H).
2 结果与讨论
2.1 反应条件优化

图5 转氨化条件的筛选
合成得到吡哆醛类催化剂9a-g和12后,对α-酮酸转氨化的反应条件进行了考察(表1).以萘乙基酮酸4a为底物,分别对催化剂、溶剂和氨源等进行了筛选.没有催化剂的情况下,只有微量产物生成,说明催化剂对转氨化反应至关重要(表1,Entry 1).催化剂9a-g和12都表现出一定的催化活性(表1,Entries 2~9),都有相应的氨基酸2a生成,其中催化剂12最好,转氨反应收率高达90%.对反应溶剂对反应有较大的影响(表1,Entries 10~16),二氯甲烷中几乎没有反应,可能是由于二苯基甘氨酸在二氯甲烷中的溶解度不好(表1,Entry 13).其他的溶剂中转氨化都有反应,其中以甲醇为溶剂时分离收率最高,因此选用甲醇-水为反应的溶剂.对氨源的筛选表明苄胺13b-c(表1,Entries 17~18)和氨基酸类化合物13a和13d-g(表1,Entries 9 and 19~22)都可以用作该反应的氨源,其中当以二苯基甘氨酸[61-67](13a)为氨源时,反应具有最高的分离收率(表1,Entry 9).

表1 转氨反应条件的筛选a
aAll reactions were carried out with keto acid 4a(0.050 mmol for entries 1-9 and 0.10 mmol for entries 10-22),2,2-diphenyl amino acids 19 (0.051 mmol for entries 1-9 and 0.101 mmol for entries 10-22),catalyst (0.010 mmol),solvent (0.50 mL) at 25 ℃ for 2 d for entries 1-9 and 3 d for entries 10-22.bIsolated yield based on keto acid 4a.
2.2 底物的拓展
在优化的反应条件下,对α-酮酸底物进行了拓展.如图6所示,所考察的α-酮酸都有一定的反应活性,芳香族类的α-酮酸(图6 2a,2c-d,2h-j)在催化剂的作用下表现出较高的反应活性,转氨化产物收率较高;脂肪族类的α-酮酸转氨化产物收率相对较低(图6 2b,2e,2k,2l).底物的立体位阻对反应有一定的影响,β-取代的酮酸(图6 2l)和β-芳基酮酸(图6 2m)在该反应中表现出较低的活性.
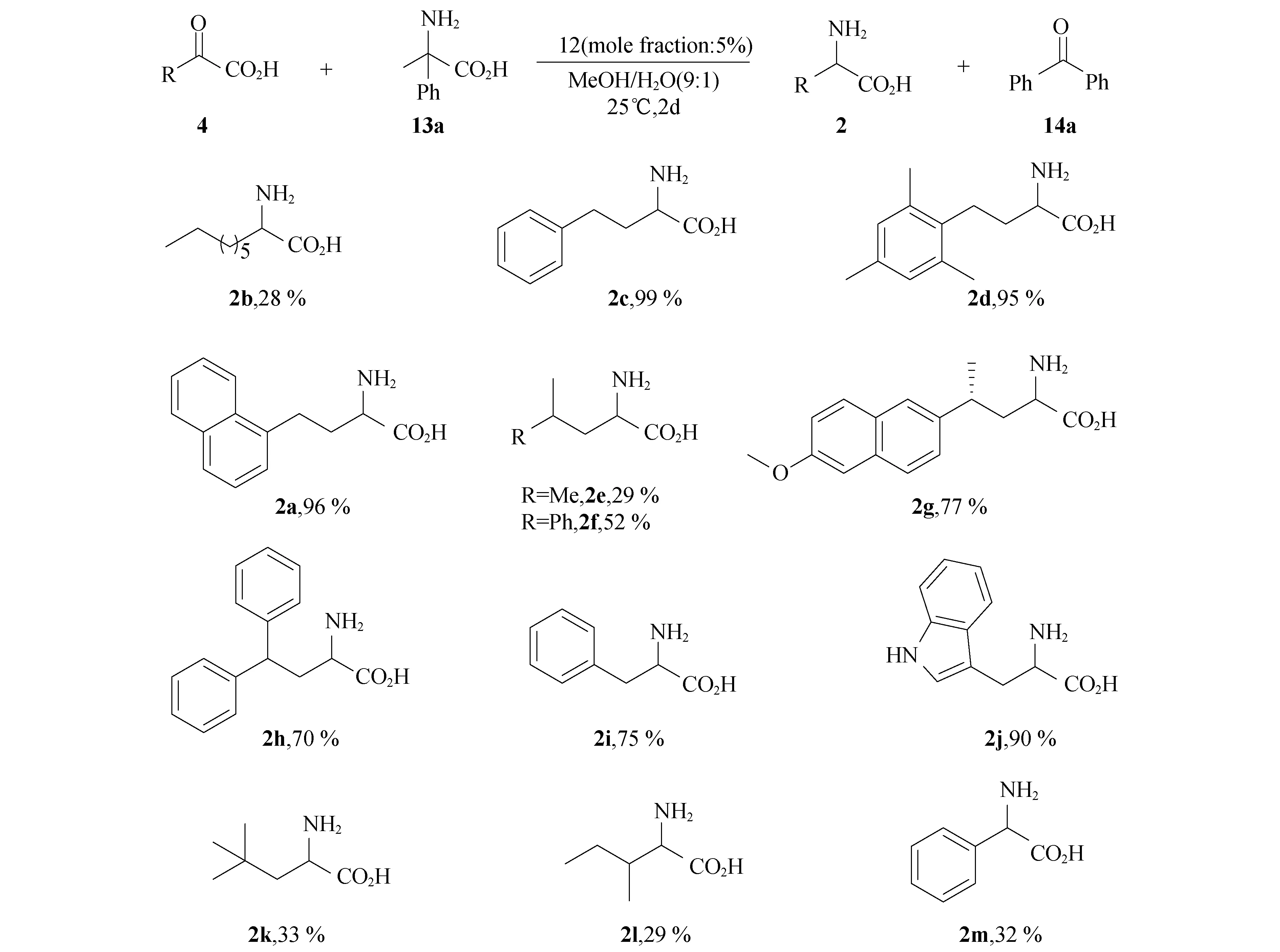
图6 底物拓展a
aAll reactions were carried out with α-keto acid 4 (0.20 mmol),α,α-diphenylglycine (13a)(0.21 mmol,and catalyst 12(0.010 mmol) in methanol (0.90 mL)and water (0.10 mL) at 25 ℃ for 2 days.bIsolated yield based on keto acidα-keto acid 4.
2.3 转氨反应机理
对上述的转氨化反应,提出了如图7所示的反应机理.首先,吡哆醛催化剂12与体系中的2,2-二苯基氨基酸(13a)缩合生成席夫碱15,席夫碱15经脱羧、转氨化和水解生成吡哆胺18;化合物18与α-酮酸4缩合形成酮亚胺19,酮亚胺19的苄位去质子,得到一个离域的碳负离子中间体20,中间体20的羧基α位质子化形成醛亚胺21,再水解释放出自由的α-氨基酸2,同时再生吡哆醛催化剂12,完成一个催化循环[63-67].
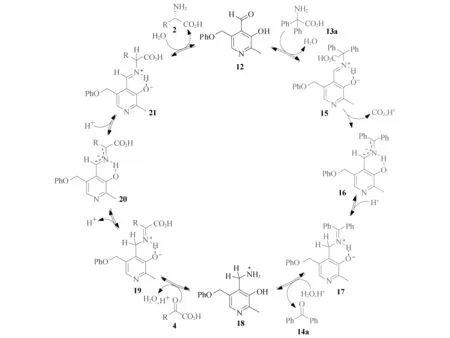
图7 吡哆醛催化α-酮酸转氨化的反应机理
3 结 论
通过在吡哆醛的侧链引入不同的官能团,合成了一系列的吡哆醛催化剂9a-g和12,该吡哆醛可以用于催化α-酮酸的转氨化,生成一系列α-氨基酸化合物.该反应条件温和,操作简单,是一种新的氨基酸合成方法.该转化还模拟了生物体系内酶催化转氨化过程,为该生物过程的理解提供了有用的信息,具有一定的化学生物学意义.
[1] 李良铸,李明晔.生化制药剂学 [M].北京:中国医药科技出版社,1991.
[2] Zhu D,Hua L.Biocatalytic asymmetric amination of carbonyl functional groups-a synthetic biology approach to organic chemistry [J].Biotechnology Journal,2009,4(4):1420-1431.
[3] Ward J,Wohlgemuth R.High-yield biocatalytic amination reactions in organic synthesis [J].Current Organic Chemistry,2010,14(17):1914-1927.
[4] Koszelewski D,Tauber K,Faber K,et al.ω-Transaminases for the synthesis of non-racemic α-chiral primary amines [J].Trends in Biotechnology,2010,28(6):324-332.
[5] Breslow R.Biomimetic chemistry and artificial enzymes:catalysis by design [J].Accounts of Chemical Research,1995,28(3):146-153.
[6] Murakami Y,Kikuchi J I,Hisaeda Y,et al.Artificial enzymes [J].Chemical Reviews,1996,96(2):721-758.
[7] Han J,Sorochinsky A E,Ono T,et al.A.Biomimetic transamination-a metal-free alternative to the reductive aminat [J].Current Organic Synthesis.2011,8(2):281-294.
[8] So S M,Kim H,Mui L,et al.Mimicking nature to make unnatural amino acids and chiral diamines [J].European Journal of Organic Chemistry,2012,2012(2):229-241.
[9] Xie Y,Pan H,Liu M,et al.Progress in asymmetric biomimetic transamination of carbonyl compounds [J].Chemical Society Reviews,2015,44(7):1740-1748.
[10] Metzler D E,Snell E E.Some transamination reactions involving vitamin B6 [J].Journal of the American Chemical Society,1952,74(4):979-983.
[11] Metzler D E,Ikawa M,Snell E E.A general mechanism for vitamin B6-catalyzed reactions [J].Journal of the American Chemical Society,1954,76(3):648-652.
[12] Matsuo Y.Pyridoxal catalysis of non-enzymatic transamination in ethanol solution [J].Journal of the American Chemical Society,1957,79(8),2016-2019.
[13] Bruice T C,Topping R M.Catalytic reactions involving azomethines.I.the imidazole catalysis of the transamination of pyridoxal by α-aminophenylacetic acid [J].Journal of the American Chemical Society,1963,85(10):1480-1488.
[14] Kuzuhara H,Komatsu T,Emoto S.Synthesis of a chiral pyridoxamine analog and nonenzymatic stereoselective transamination [J].Tetrahedron Letters,1978,19(38),3563-3566.
[15] Breslow R,Hammond M,Lauer M.Selective transamination and optical induction by a.beta.-cyclodextrin-pyridoxamine artificial enzyme [J].Journal of the American Chemical Society,1980,102(1):421-422.
[16] Tachibana Y,Ando M,Kuzuhara H.Asymmetric synthesis ofα-amino acids by nonenzymatic transamination.Versatility of the reaction and enantiomeric excesses of the products [J].Chemistry Letters,1982,11(11):1765-1768.
[17] Tachibana Y,Ando M,Kuzuhara H.Asymmetric synthesis ofα-amino acids by nonenzymatic transamination.Rationalization for the stereochemical results [J].Chemistry Letters,1982,11:1769-1772.
[18] Breslow R,Czarnik A W.Transaminations by pyridoxamine selectively attached at C-3 in.beta.-cyclodextrin [J].Journal of the American Chemical Society,1983,105(5):1390-1391.
[19] Zimmerman S C,Czarnik A W,Breslow R.Intramolecular general base-acid catalysis in transaminations catalyzed by pyridoxamine enzyme analogs [J].Journal of the American Chemical Society,1983,105(6):1694-1695.
[20] Tachibana Y,Ando M,Kuzuhara H.Asymmetric synthesis ofα-deuteratedα-amino acids through nonenzymatic transamination reaction and the determination of their enantiomeric excesses [J].Bulletin of the Chemical Society of Japan,1983,56(12):3652-3656.
[21] Zimmerman S C,Breslow R.Asymmetric synthesis of amino acids by pyridoxamine enzyme analogs utilizing general base-acid catalysis [J].Journal of the American Chemical Society,1984,106(5):1490-1491.
[22] Breslow R,Czarnik A W,Lauer M,et al.Mimics of transaminase enzymes [J].Journal of the American Chemical Society,1986,108(8):1969-1979.
[23] Breslow R,Chmielewsk J,Foley D,et al.Optically active amino acid synthesis by artificial transaminase enzymes [J].Tetrahedron,1988,44(17):5515-5524.
[24] Ando M,Kuzuhara H.Synthesis and reaction of a new chiral pyridoxamine analogue;Some doubt about the stereochemical process tentatively proposed for a nonenzymatic transamination reaction [J].Bulletin of the Chemical Society of Japan,1989,62(1):244-250.
[25] Ando M,Kuzuhara H.Asymmetric synthesis of fluorophenylalanines and (trifluoromethyl) phenylalanines;the use of chiral pyridoxamine-like pyridinophane-zinc complex as an enzyme mimic [J].Bulletin of the Chemical Society of Japan,1990,63(7):1925-1928.
[26] Fasella E,Dong S D,Breslow R.Reversal of optical induction in transamination by regioisomeric bifunctionalized cyclodextrins [J].Bioorganic & Medicinal Chemistry,1999,7(5):709-714.
[27] Zhou W,Yerkes N,Chruma J J,et al.Chiral polyamines from reduction of polypeptides:asymmetric pyridoxamine-mediated transaminations [J].Bioorganic & Medicinal ChemistryLetters,2005,15(5):1351-1355.
[28] Bandyopadhyay S,Zhou W,Breslow R.Isotactic polyethylenimines induce formation of l-amino acids in transamination [J].Organic Letters,2007,9(6):1009-1012.
[29] Breslow R,Wei S,Kenesky C.Enantioselective transaminations by dendrimeric enzyme mimics [J].Tetrahedron,2007,63(27):6317-6321.
[30] Wei S,Wang J,Venhuizen S,et al.Dendrimers in solution can have their remote catalytic groups folded back into the core:Enantioselective transaminations by dendritic enzyme mimics-II [J].Bioorganic & Medicinal Chemistry Letters,2009,19(19):5543-5546.
[31] Bernauer K,Deschenaux R,Taura T.Stereoselectivity in reactions of metal complexes VII.Asymmetric synthesis of amino acids by metal ion-promoted transamination [J].Helvetica Chimica Acta,1983,66(7):2049-2058.
[32] Deschenaux R,Bernaue K.Stereoselectivity in reactions of metal complexes VIII.Asymmetric synthesis of some amino acids by stereoselective transamination of aliphatic keto acids in mixed ligand copper(II)-Schiff-base complexes [J].Helvetica Chimica Acta,1984,67(2):373-377.
[33] Svenson J,Zheng N,Nicholls I A.A molecularly imprinted polymer-based synthetic transaminase [J].Journal of the American Chemical Society,2004,126(27):8554-8560.
[34] Kikuchi J I,Zhang Z Y,Murakami Y.Enantioselective catalysis by supramolecular bilayer membrane as artificial aminotransferase [J].Chemistry Letters,1994,23(8):1559-1562.
[35] Kikuchi J I,Zhang Z Y,Murakami Y.Enantioselective catalysis by a supramolecular bilayer membrane as an artificial aminotransferase.stereochemical roles of an L-lysine residue and L-phenylalanine at the reaction site [J].Journal of the American Chemical Society,1995,117(19):5383-5384.
[36] Kuang H,Brown M L,Davies R R,et al.Enantioselective reductive amination of α-keto acids to α-amino acids by a pyridoxamine cofactor in a protein cavity [J].Journal of the American Chemical Society,1996,118(44):10702-10706.
[37] Kuang H,Distefano M D.Catalytic enantioselective reductive amination in a host-guest system based on a protein cavity [J].Journal of the American Chemical Society,1998,120(5):1072-1073.
[38] Qi D,Kuang H,Distefano M D.Effects of metal ions on the rates and enantioselectivities of reactions catalyzed by a series of semisynthetic transaminases created by site directed mutagenesis [J].Bioorganic & Medicinal Chemistry Letters,1998,8(7):875-880.
[39] Häring D,Distefano M D.Enzymes by design:Chemogenetic assembly of transamination active sites containing lysine residues for covalent catalysis [J].Bioconjugate Chemistryistry,2001,12(3):385-390.
[40] Soloshonok V A,Kirilenko A G,Galushko S V,et al.Catalytic asymmetric synthesis of β-fluoroalkyl-β-amino acids via biomimetic[1,3]-proton shift reaction [J].Tetrahedron Letters,1994,35(28):5063-5064.
[41] Willems J G H,Vries J G,Nolte R J M,et al.Asymmetric imine isomerisation in the enantioselective synthesis of chiral amines from prochiral ketones [J].Tetrahedron Letters,1995,36(22):3917-3920.
[42] Soloshonok V A,Ono T.Highly enantioselective transfer of chirality from a less to a more configurationally unstable stereogenic center.A Practical asymmetric synthesis of (Fluoroalkyl) amines via biomimetic transamination [J].Journal of Organic Chemistry,1997,62(10):3030-3031.
[43] Soloshonok V A,Ono T,Soloshonok I V.Enantioselective biomimetic transamination of β-keto carboxylic acid derivatives.an efficient asymmetric synthesis of β-(Fluoroalkyl) β-amino acids [J].Journal of Organic Chemistry,1997,62(22):7538-7539.
[44] Xiao J,Zhang X,Yuan C.A facile asymmetric synthesis of 1-amino-2,2,2-trifluoroethanephosphonic acid [J].Heteroatom Chemistry,2000,11(7):536-540.
[45] Hjelmencrantz A,Berg U.New Approach to biomimetic transamination using bifunctional[1,3]-proton transfer catalysis in thioxanthenyl dioxide imines [J].Journal of Organic Chemistry,2002,67(11):3585-3594.
[46] Knudsen K R,Bachmann S,Jørgensen K A.Catalytic enantioselective transamination of α-keto esters:An organic approach to enzymatic reactions [J].Chemical Communications,2003,35(20):2602-2603.
[47] Bachmann S,Knudsen K R,Jørgensen K A.Mimicking enzymatic transaminations:Attempts to understand and develop a catalytic asymmetric approach to chiral α-amino acids [J].Organic & Biomolecular Chemistry2004,2(14):2044-2049.
[48] Soloshonok V A,Yasumoto M.Catalytic asymmetric synthesis of α-(trifluoromethyl)benzylamine via cinchonidine derived base-catalyzed biomimetic 1,3-proton shift reaction [J].Journal of Fluorine Chemistry,2007,128(3):170-173.
[49] Xiao X,Xie Y,Su C,et al.Organocatalytic asymmetric biomimetic transamination:From α-keto esters to optically active α-amino acid derivatives [J].Journal of the American Chemical Society,2011,133(4):12914-12917.
[50] Wu Y,Deng L.Asymmetric synthesis of trifluoromethylated amines via catalytic enantioselective isomerization of imines [J].Journal of the American Chemical Society,2012,134(10):14334-14337.
[51] Xie Y,Pan H,Xiao X,et al.Organocatalytic asymmetric biomimetic transamination of aromatic ketone to optically active amine [J].Organic & Biomolecular Chemistry,2012,10(45):8960-8962.
[52] Xue F,Xiao X,Wang H,et al.The effect of benzyl amine on the efficiency of the base-Catalyzed Transamination of α-Keto Esters [J].Tetrahedron,2012,68(34):6862-6867.
[53] Xiao X,Liu M,Rong C,et al.An efficient asymmetric biomimetic transamination of α-keto esters to chiral α-amino esters [J].Organic Letters,2012,14(20):5270-5273.
[54] Liu M,Li J,Xiao X,et al.An efficient synthesis of optically active trifluoromethyl aldimines via asymmetric biomimetic transamination [J].Chemical Communications,2013,49(21):1404-1406.
[55] Pan H,Xie Y,Liu M,et al.Organocatalytic asymmetric biomimetic transamination of α-keto acetals to chiral α-amino acetals [J].Rsc Advances,2014,4(5):2389-2392.
[56] Su C,Xie Y,Pan H,et al.Organocatalytic synthesis of optically active β-branched α-amino esters via asymmetric biomimetic transamination [J].Organic & Biomolecular Chemistry,2014,12(31):5856-5860.
[57] Shi L,Yang Q,Tao C,et al.Chiral pyridoxal-catalyzed asymmetric biomimetic transamination of α-keto acids [J].Organic Letters,2015,17(23):5784-5787.
[58] Lan X,Tao C,Liu X,et al.Asymmetric transamination of α-keto acids catalyzed by chiral pyridoxamines [J].Organic Letters,2016,18(15):3658-3661.
[59] Liu Y E,Lu Z,Li B,et al.Enzyme-inspired axially chiral pyridoxamines armed with a cooperative lateral amine chain for enantioselective biomimetic transamination [J].Journal of the American Chemical Society,2016,138(34):10730-10733.
[60] Chen J,Zhao J,Gong X,et al.A new type of chiral-pyridoxamines for catalytic asymmetric transamination of α-keto acids [J].Tetrahedron Letters,2016,57(41):4612-4615.
[61] Liu L,Zhou W,Chruma J,et al.Transamination reactions with multiple turnovers catalyzed by hydrophobic pyridoxamine cofactors in the presence of polyethylenimine polymers [J].Journal of the American Chemical Society,2004,126(26):8136-8137.
[62] Chruma J J,Liu L,Zhou W,et al.Hydrophobic and electronic factors in the design of dialkylglycine decarboxylase mimics [J].Bioorganic & Medicinal Chemistry,2005,13(20):5873-5883.
[63] Ding L,Chen J,Hu Y,et al.Aminative umpolung of aldehydes to α-amino anion equivalents for Pd-catalyzed allylation:An efficient synthesis of homoallylic amines [J].Organic Letters,2014,16(29):720-723.
[64] Liu X,Gao A,Ding L,et al.Aminative umpolung synthesis of aryl vicinal diamines from aromatic aldehydes [J].Organic Letters.2014,16(8):2118-2121.
[65] Xu J,Chen J,Yang Q,et al.Synthesis of α-methylidene-γ-amino acid esters from aldehydes via an aminative umpolung strategy [J].Advanced Synthesis & Catalysis,2014,356(14-15):3219-3224.
[66] Wu L,Xie C,Mei H,et al.Synthesis of trifluoromethyl-containing vicinal diamines by asymmetric decarboxylative mannich addition reactions [J].Journal of Organic Chemistry,2015,80(6):3187-3194.
[67] Tang S,Park J Y,Yeagley A A,et al.Decarboxylative generation of 2-azaallyl anions:2-iminoalcohols via a decarboxylative erlenmeyer reaction [J].Organic Letters,2015,17(9):2042-2045.
(责任编辑:郁 慧,包震宇)
Transamination of α-keto acids catalyzed by pyridoxals
CHEN Jing, ZHAO Junyu, ZHAO Baoguo
(College of Life and Environment Sciences,Shanghai Normal University,Shanghai 200234,China)
In this paper,we designed and synthesized a series of pyridoxals tagged with different functional groups.The pyridoxals exhibited high catalytic activity in transamination of α-keto acids to obtain various amino acids in good yields.This transamination has some obvious advantages such as mild reaction conditions and easy operation.This workhas provided a potentially practical method for the synthesis of chemically and biologically significant α-amino acids.
α-keto acid; transamination; α-amino acid; pyridoxal
2016-09-22
国家自然科学基金 (21272158,21672148).
赵宝国,中国上海市徐汇区桂林路100号,上海师范大学生命与环境科学学院,邮编:200234,E-mail:zhaobg2006@hotmail.com
O 62
A
1000-5137(2016)06-0719-10
猜你喜欢
中华养生保健(2020年2期)2020-11-16 00:49:20
云南医药(2020年5期)2020-10-27 01:37:50
河南畜牧兽医(2020年11期)2020-01-11 05:31:49
医学新知(2019年4期)2020-01-02 11:04:02
西安文理学院学报(自然科学版)(2016年4期)2016-12-19 08:18:59
China International Studies(2016年3期)2016-07-14 03:00:06
兽医导刊(2016年6期)2016-05-17 03:50:53
农村农业农民·B版(2015年9期)2015-10-16 16:11:41
质谱学报(2015年5期)2015-03-01 03:18:25
云南中医学院学报(2014年2期)2014-11-07 02:48:12
