
ABI PGM测序平台用于细菌基因组de novo测序的评价
2016-01-15 02:02:00黄方亮
生物信息学 2015年2期
ABI PGM测序平台用于细菌基因组de novo测序的评价
黄方亮
(浙江大学生命科学学院大型仪器平台,杭州310058)
摘要:为了探索加快细菌基因组研究的方法,利用ABI PGM 测序平台测定了 1 株单细胞硫还原地杆菌的基因组序列。测序共获得1.4 Gbp 数据,平均读长为177 bp。通过多个拼接软件并采用合适的组装策略,得到一个完整细菌基因组3.55 Mbp和一条完整质粒序列110 kbp。测定基因组序列与参考基因组kn400序列的相似性达到 94%,参考基因组91%的基因能在测定基因组中找到相似基因。通过本研究表明采用ABI PGM测序平台结合灵活的拼接策略可快速构建细菌基因组精细图谱,为进一步的功能注释及深入的信息分析提供准确的数据,大大加快研究进程。
关键词:PGM测序平台;细菌基因组测序
中图分类号:Q75文献标志码:A
收稿日期:2015-04-04;修回日期:2015-04-20.
基金项目:国家自然科学
作者简介:单光宇,男,硕士研究生,研究方向:生物信息学;E-mail: sci@shanguangyu.com.
doi:10.3969/j.issn.1672-5565.2015.02.08
Evaluation of PGM sequencing platform using in bacterial genome de novo sequencing
HUANG Fangliang
(EquipmentandTechnologyServicePlatformofCollegeofLifeSciencesZhejianguniversity,Hangzhou310058,China)
Abstract:In order to speed up bacterial genome exploration, we performed the genome sequencing of Geobacter sulfurreducens using PGM. Totally, 1.4 Gbp raw data were obtained with an average read length of 177 bp. 2 contigs were assembled by multiple software calculations using appropriate assembly strategies. The size of whole obtained genome and plasmid was measured to be 3.55 Mbp and 110 kbp, respectively. The sequenced genome identified 94% of reference genome strain KN400 and 91% genes of KN400 were tested to be orthologous in the sequenced genome. This study proved that the use of ABI PGM sequencing platform with splicing flexible strategy can rapidly build bacteria genome map. By providing accurate data for the functional annotation and in-depth information analysis, it will greatly accelerate research progress.
Keywords:ABI PGM Sequencing Platform; Bacterial Genome de novo Sequencing
随着测序技术的迅速发展和测序成本的急速降低,细菌全基因组精细测序成为科学家研究目的细菌的基本要求[1]。2005年罗氏454测序仪出现后,一次开机产生上百万条数据的高通量测序技术大大加快了基因组研究的进程[2],2012年454测序仪发明人Jonathan Rothberg 博士在焦磷酸测序[3]的基础上,发明了新一代测序仪ABI PGM,它的测序通量更有弹性,能够使用314、316、318三种芯片,分别出10 M,100 M,1 G测序数据。用半导体检测技术替代了冷光CCD拍照成像技术检测DNA信号,测序成本更低,原始数据占用的计算机资源更少[4]。一张芯片上机测序只要3小时。利用ABI PGM 318芯片配合本来用于5500测序仪上的mate pair试剂盒,使ABI PGM测序平台成为细菌基因组精细测序的强大工具。
本研究中,我们希望快速得到目的菌株完整基因组序列。为此,构建了200 bp短片段文库和3 KB mate pair文库,接上不同的接头,使用PGM测序。得到的数据用CLC Bio Genomics work bench 6.0(CLC Bio, Aarhus, Denmark)软件拼接,采用合适的拼接策略后,两周左右就得到完整的目的细菌基因组精细图谱。
1材料与方法
1.1菌株培养和核酸提取
单细胞硫还原地杆菌菌株由浙大热能所提供,挑取单克隆菌落,在37 ℃下用改进过的LB液体培养基密闭振荡培养过夜。取200 mL菌液最高速离心1 min,弃上清,将沉淀转入研钵,加液氮研磨,研磨充分后加入 1 mL Plant DNAzol ,2 μL 2-ME( β-巯基乙醇)继续研磨,转移裂解产物至1.5 mL离心管中。将离心管置65 ℃水浴 30 min。加750 μL氯仿,混合均匀。12 000 rpm,离心5 min。小心取上清(避免吸取中间蛋白层),转入一新的1.5 mL 管(体积大约有600 μL)。加0.7体积的异丙醇(约420 μL),12 000 rpm,离心10 min。弃上清,加入1 mL 75%乙醇至离心管中,颠倒数次以重悬DNA,直立离心管1 min至DNA团块沉至管底,倾去或吸除洗涤液。细小的DNA沉淀团块容易在倾倒洗涤液时丢失,可室温3 000 rpm,离心3 ~ 5 min,然后倾去或吸除洗涤液。重复清洗1次。最后简短离心,用枪头小心吸弃残留液体。室温静置数分钟(约10 min)使残余乙醇挥发,注意不要完全晾干DNA。加入适量(100 ~ 200 μL)灭菌双蒸水或TE缓冲液,使DNA 沉淀溶解。向DNA溶液中加入终浓度为40 μg·mL-1的RNase A,37 ℃孵育 30 min,-20 ℃保存。
1.2基因组测序文库构建及PGM测序
取200 ng目的细菌基因组DNA,用millipore水稀释到50 μL体积,放入Biorupt,参数:Power Level:L ,Time ON:0.5 min,Time OFF:0.5 min,Number of 15-min Cycles:3。超声破碎到250 bp左右,用Ion XpressTMPlus Fragment Library Kit构建200 bp左右测序文库。取3 μg基因组DNA,用millipore水稀释到150 μL体积,利用hydroshear核酸片断化仪打断到3 KB,参数:Standard Shearing Assembly,SC 13,20cycles。1%凝胶电泳回收纯化,使用5500 SOLID MATE-PAIRED LIBRARY KIT构建3 KB mate-pair文库。两个文库接不同的接头,上PGM测序,PGM测序参照ABI PGM操作手册。
1.3测序数据de novo拼接
将两个文库数据导入CLC Bio Genomics work bench 6.0,用trimed功能去除低质量数据后,以de novo模块拼接。参数使用如下:word size values 范围是25~40核苷酸,bubble sizes 选择 50 bp, 60 bp,70 bp三种,Map reads back to contig(slow):mismatch cost:2,insertion cost:2,deletion cost:3,length fraction:0.5,similarity fraction:0.8。将得到的最理想拼接结果做为参照序列,比对得到的两个文库数据,从而填补scaffold序列中的gap,并根据落在两个不同scaffold上的成对mate-pair数据,确认scaffold间的关系。不同参数条件拼接出来的contigs重新mapping回拼好的scaffold上,消除gap。拼接策略见图1。
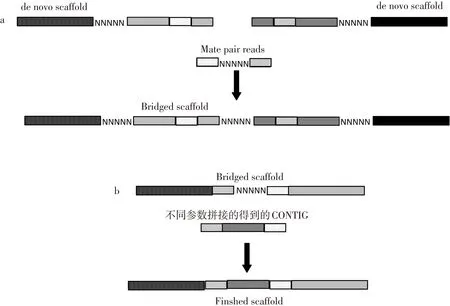
图1 拼接策略示意图
注:(a) 利用3 KB mate pair数据确定scaffold间关系; (b) 利用不同参数条件下得到的contig序列,填补scaffold中的gap,得到完整序列。
Notes:(a) Scaffold ordering phase:using 3 KB mate pair data to determine the relationship between scaffolds; (b) Genome finish phase:fill gap by contig mapping.
1.4基因组FINISH
经1.3拼接后,得到成环的基因组序列,根据缺少的gap,设计基于gap的引物。经PCR扩增后,利用一代测序仪3130的数据,补全序列,从而构建完整环状基因组。
1.5基因预测注释分析
将基因组数据提交到RAST(Rapid Annotation using Subsystem Technology)[5]网站,得到3 822个预测基因。结合另外几个原核生物基因预测软件Glimmer[6],Genemarker[7],FgeneSB[8]校正预测结果。利用RAST网站Compare模块中的function based功能与其它基因组做功能比较。KEGG模块看基因组中基因所在pathway信息。并与InterPro[9],COG[10]数据库比对确认预测基因生化代谢功能。对于非蛋白质编码基因rRNA和tRNA的预测,分别用RNAmmer[11]和tRNAscanSE[12]确认。
1.6基因组比较分析
选取单细胞硫还原地杆菌生物型菌株kn400[13]做为参考序列,运用NCBI网站的Blast2SEQ软件比较两个基因组相似性。根据预测的基因,用RAST网站的compare基于sequence based查找参考基因组中的同源基因。
2结果
2.1测序数据量和基因组拼接
两个文库共获得8.1 M条序列,1.4 Gbp碱基,数据详情见表1。将数据导入CLC分析软件,经过trimed后,还有7.8 M条序列可用,序列统计见图2。经过多次de novo拼接,调整各种参数,最后word size values选35, bubble sizes选择60 bp,组装成16个scaffolds,总长3.66 M,N50为492 k,最大长度889 k。将16个scaffolds序列做为参照序列,把两个文库的数据mapping上去,找到16个scaffolds间的前后关系,并补上scaffold中的gap。经过多轮的mapping最终将基因组拼接成一个环状染色体序列3.55 M,并发现一个完整的质粒序列110 KB,基因组G+C含量61%。环状染色体序列中还有4个不能通过序列拼接确定的gap,用PRIMER3在线软件设计引物[14],PCR扩增测序后,拼回原来的位置得到一个完整的环状染色体序列,扩增产物电泳图见图3。

表1 200 bp及3 KB mate pair文库数据统计情况
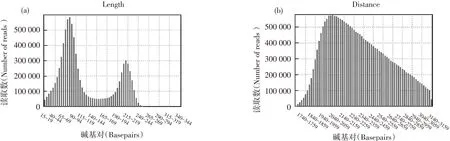
图2 序列示意图
注:(a) 200 bp文库和3 KB mate pair文库序列读长分布; (b) 3 KB mate pair数据在基因组上的实际定位统计,峰值出现在2.1 KB,范围在1.7 KB~3.1 KB间。
Notes:(a) Read length distribution of the 200 bp library and 3 KB mate pair library; (b) Distance of 3 KB mate pair library data locate in genome, peak appeared in the 2.1 KB, ranging from between 1.7 KB~3.1 KB.
2.2与参考序列比较结果
选择基因组大小为3.7 M 的kn400做为参考,进行基因组比对,结果显示两个基因组序列相似度94%。参考基因组中91%的基因能在测定基因组的预测基因中找到,相似度≥95%的基因占52%,95%>相似度≥30%的基因占39%。
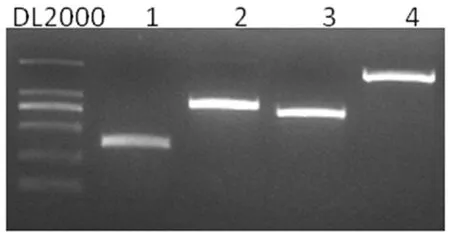
图3 电泳图
注:1,2,3,4分别是四个gap PCR产物电泳条带。
Notes:1~4 is PCR amplification products of 4 gaps.
3讨论
目前,得到细菌全基因组序列完整图谱已经是高质量细菌文章发表的必备条件。而很多时候科学家在高通量测序完成后,得到的是几十个独立的scaffolds,要找到它们之间的关系,拼接成环状完整的基因组,还需要订购很多的引物,几个月的时间做PCR扩增,费时费力。采用200 bp文库加3 KB mate pair文库,用PGM 318芯片测序后,得到1.4 G原始数据,经过高质量筛选后,余下881 M数据,覆盖基因组266倍左右,软件初步拼接得到16个scaffolds。将16个scaffolds做为参考序列,把所有测序数据mapping上去,通过定位在两个不同scaffolds上的多个成对的mate-pair序列来确定scaffolds间的前后关系,也可以结合软件SSPACE来辅助确认scaffolds间的关系。确认关系排好顺序的scaffolds被拼接到一起,做为参考序列,再做mapping,通过mapping结果可以进一步确认是否正确拼接scaffolds。如此反复,直到拼接成环状序列。过程中可以结合gap修复软件Gapfiller[15],SOAPdenovo GapCloser v1.12r6来关闭gaps[16]。可能是因为重复序列的关系,环状基因组中还是会有4个gap无法修复,最终通过设计引物PCR扩增,3130测序,拼接出完整的基因组数据。拼接完成后还检测到一个完整的质粒序列。
PGM测序平台还应用到了另外几个细菌基因组的研究中,都得到完整的细菌基因组图谱。但经过实验发现如果目的细菌中出现多个质粒,且质粒间的序列高度相似时,虽然可以得到完整的基因组数据,却很难保证得到完整的质粒序列。必须将质粒分离开单独测序才行。本研究实验结果证明PGM单次上机成本较低,一天就能完成两张318芯片测序,一张318芯片数据足够满足4 M左右细菌基因组的精细图拼接。因此采用ABI PGM测序平台结合合适的拼接软件,采用灵活的拼接策略可以快速构建细菌基因组精细图谱,为进一步的基因功能注释和深入的信息分析提供准确的数据,能够大大加快细菌基因组研究的进程。
参考文献(References)
[1]BARBOSA E G, ABURJAILE F F, RAMOS R T, et al. Value of a newly sequenced bacterial genome[J]. World J Biol Chem,2014, 5(2): 161-168.
[2]YANG Y, XIE B, YAN J. Application of next-generation sequencing technology in forensic science[J]. Genomics Proteomics Bioinformatics, 2014, 12(5): 190-197.
[3]RONAGHI M, UHLEN M, NYREN P. A sequencing method based on real-time pyrophosphate[J]. Science, 1998, 281(5375): 363-365.
[4]MERRIMAN B, ROTHBERG J M. Progress in ion torrent semiconductor chip based sequencing[J]. Electrophoresis, 2012, 33(23): 3397-3417.
[5]OVERBEEK R, OLSON R, PUSCH G D, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST)[J]. Nucleic Acids Res, 2014, 42(Database issue): 206-214.
[6]DELCHER A L, BRATKE K A, POWERS E C, et al. Identifying bacterial genes and endosymbiont DNA with Glimmer[J]. Bioinformatics, 2007, 23(6): 673-679.
[7]HOLLAND M M, PARSON W. GeneMarker(R) HID: A reliable software tool for the analysis of forensic STR data[J]. J. Forensic Sci, 2011, 56(1): 29-35.
[8]VICTOR S, ASAF S. Automatic annotation of microbial genomes and metagenomic sequences in metagenomics and its applications in agriculture[J]. Biomedicine and Environmental Studies, 2011: 61-78.
[9]HUNTER S, JONES P, MITCHELL A, et al. InterPro in 2011: new developments in the family and domain prediction database[J]. Nucleic Acids Res, 2012, 40(Database issue): D306-312.
[10]TATUSOV R L, KOONIN E V, LIPMAN D J. A genomic perspective on protein families[J]. Science, 1997, 278(5338): 631-637.
[11]LAGESEN K, HALLIN P, RODLAND E A, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes[J]. Nucleic Acids Res, 2007, 35(9): 3100-3108.
[12]SCHATTNER P, BROOKS A N, LOWE T M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs[J]. Nucleic Acids Res, 2005, 33(Web Server issue): W686-689.
[13]BUTLER J E, YOUNG N D, AKLUJKAR M, et al. Comparative genomic analysis of Geobacter sulfurreducens KN400, a strain with enhanced capacity for extracellular electron transfer and electricity production[J]. BMC Genomics, 2012, 13: 471.
[14]UNTERGASSER A, CUTCUTACHE I, KORESSAAR T, et al. Primer3-new capabilities and interfaces[J]. Nucleic Acids Res, 2012, 40(15): e115.
[15]NADALIN F, VEZZI F, POLICRITI A. GapFiller: a de novo assembly approach to fill the gap within paired reads[J]. BMC Bioinformatics, 2012, 13 (Suppl 14): S8.
[16]LUO R, LIU B, XIE Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler[J]. Gigascience, 2012, 1(1): 18.
*通信作者:卢一鸣,男,军事医学科学院助理研究员,研究方向:生物信息学;E-mail: luym@outlook.com.
张成岗,男,研究员,博士生导师,研究方向:生物信息学;E-mail:zhangcg@bmi.ac.cn.
猜你喜欢
现代实用医学(2022年10期)2022-12-08 05:50:08
今日农业(2021年11期)2021-08-13 08:53:24
中国民间疗法(2021年9期)2021-07-22 08:05:52
猪业科学(2021年3期)2021-05-21 02:05:36
幽默大师(2020年10期)2020-11-10 09:07:22
中华诗词(2019年1期)2019-11-14 23:33:56
猪业科学(2018年4期)2018-05-19 02:04:31
组合机床与自动化加工技术(2014年12期)2014-03-01 02:22:47
遗传(2014年3期)2014-02-28 20:58:49
无机化学学报(2014年5期)2014-02-28 17:31:34
