
基于EST数据的水稻基因表达大规模初步分析
2016-01-15 02:01:52宋东光
生物信息学 2015年2期
关键词:水稻
基于EST数据的水稻基因表达大规模初步分析
宋东光
(佛山科学技术学院园艺系,广东 佛山 528231)
摘要:EST序列代表了组织基因表达的转录信号,本研究尝试开发简单高效的大规模EST分析方法,从NCBI下载水稻(Oryza sativa) 的所有EST序列并进行分析以获取水稻发育过程基因表达的重要信息。通过进行blast比对和phrap拼接分析,及利用Unix文本过滤方法,从EST序列拼接获得了3万多个重叠群序列。进一步将重叠群序列与NCBI核酸数据库进行比对获得了各个序列的注释信息。从重叠群的组织表达初步挖掘中发现花药的表达数量最多,为下一步探讨水稻发育器官特异表达基因调控打下了重要基础。
关键词:水稻;EST;Bast; Phrap; 组织特异表达
中图分类号:Q344+.13文献标志码:A
收稿日期:2015-01-19;修回日期:2015-04-24.
基金项目:国家自然
作者简介:杨红,女,讲师,研究方向:应用数学,生物信息学;E-mail: yanghong19820118@163.com.
doi:10.3969/j.issn.1672-5565.2015.02.05
Large-scale preliminary analysis of rice gene expression mining from EST data
SONG Dongguang
(DepartmentofHorticulture,FoshanUniversity,FoshanGuangdong528231,China)
Abstract:EST sequences represent transcribed signals of gene expressions in tissues. In this study, a simple and effective method for large-scale EST analysis was developed using all rice(Oryza sativa) ESTs downloaded from NCBI for mining important information in rice development. After the blast alignment, phrap contig joining, and Unix command-line filtering, over 30 000 contigs were obtained from EST sequences. Annotations of these contigs were returned with further alignments to NCBI nucleotide databases. Anther expressions showed the most abundant in this preliminary mining from annotations for different tissues. This lays an important foundation for further investigating tissue-specific regulation of gene expression in rice development.
Keywords:Oryza sativa; EST; Blast; Phrap; Tissue-specific expression
随着功能基因组学研究的广泛开展,阐明基因表达调控网络的分子机理成为了近年来分子生物学研究的主要领域之一。获得基因活动信息的方法如EST,SAGE分析,表达芯片分析等可以提供大量的基因活动信号,并进一步从获得的各种表达数据分析构建基因调控网络。其中,EST分析获得的基因表达信息真实反映了细胞内基因活动的情况,包括基因的组织特异表达情况。大量的EST序列可以从NCBI Genbank数据库获取,研究者也能够从cDNA文库进行克隆快速测序获得,面对海量的序列数据需要有效的高通量分析工具才能提取出更多的基因表达谱信息并用于构建基因调控网络[1-3]。
EST序列预处理如如去除载体序列、poly(A)尾巴等对于后续分析是很必要的,涉及EST的各种分析包括转录组、重叠群拼接,基因注释,SSR及SNP多态性, ORF确定,选择性剪接,microRNA及非编码RNA分析,RNA编辑,GO查询,组织特异性表达谱分析以及构建基因调控网络等并取得了许多重要进展[4-10]。
本文开发了简单有效的工具以来自NCBI的水稻EST序列为材料进行大规模初步分析,包括进行blast比对,phrap重叠群拼接与注释,及组织特异表达分析,为水稻生长发育过程基因表达调控网络的构建奠定重要基础。
1材料与方法
1.1操作系统和文本过滤工具
操作系统为FreeBSD 10.0,由The FreeBSD Project(http://www.freebsd.org/)开发, 利用其内嵌的Unix命令如awk、sed、tr、uniq、split、 comm、paste、 join及sort等进行EST序列预处理[11]及其他文本挖掘工作。
1.2EST序列及格式转换
“gz”压缩格式的EST 序列数据从NCBI下载, 提取其中的重要信息并转换为一行,每个字段由制表符隔开。抽取每个EST序列及其id并转换为FASTA格式,序列开始及末尾的长于10nt的poly(A/C/G/T)通过前面的过滤命令进行去除。
1.3Blast比对分析
NCBI开发的blast程序blast-2.2.22-ia32-freebsd 用于 EST序列的本地blast比对分析, 每个EST序列彼此间进行相似性比对找出得分大于100的去除重复后合并其id于一行。
1.4重叠群拼接分析
phrap 程序(由Washington 大学的Phil Green开发, http://www.phrap.org)用于将相似性较高的EST序列重叠拼接获得重叠群(contig)。
1.5重叠群注释
将以上的拼接重叠群进行远程NCBI网络 blast 比对以获得重叠群的注释,每次可以进行200个重叠群(FASTA格式), 返回结果保存为“.txt”格式并只提取注释信息。
1.6组织特异表达谱分析
不同组织表达的EST可以通过比较组织表达的EST id及拼接注释后的重叠群id得到。
2结果
2.1大规模EST分析通路
本文的EST大规模分析流程图参照图1。
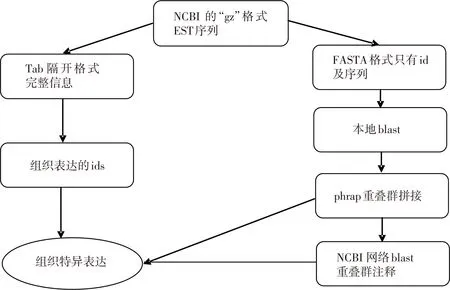
图1 本文EST大规模分析流程图(具体过程见方法)
EST序列下载后将其从“gz”格式解压缩,提取必要信息并将转换为由制表符隔开的一行数据库录入格式,含6个字段即GI-GenBank数据库中的唯一标识号, DEFINITION-EST数据定义信息,TITLE-测序记录号, /organism/-物种名, FEATURES-EST序列简单介绍, ORIGIN-EST核苷酸序列。典型的一个EST序列见图2。

图2 提取转换格式后的一条典型EST序列,含6个字段由制表符分隔,即GI, DEFINITION, TITLE,
2.2NCBI记录的不同物种EST序列统计
截止2014年2月14日从NCBI下载的所有“gz”格式的EST序列提取其GI及organism后统计了各个物种的EST总数。119个物种EST记录数超过10万条,但其中只有63个物种数量超过了20万条(见表1,只列出了部分物种)。这其中,人(Homosapiens) 和 家鼠(Musmusculus) 记录数最多,分别达到了8千7百万和4千8百多万条,排在第三位的是玉米(Zeamays)有2百多万条,水稻 (Oryzasativa)为1百多万条,包括了籼稻和粳稻(见表1)。

表1 截止2014年2月14日从NCBI下载的所有物种记录数
2.3水稻EST序列彼此间的blast比对
水稻的125万条EST序列(截止2010年3月24日,包括籼稻和粳稻)经过预处理去除了poly(A/T/G/C)后利用本地的blast程序进行了比对,比对工作连续进行约用时1个多月,之后将彼此比对打分达到100以上的序列ids(即GI号)合为一行,得到1 237 411行id组,部分示例列于图3。

图3 相似性比对(打分100及以上的)EST序列其id合为一行
blast比对是用水稻的每个EST序列与所有的EST进行两两比对得到的结果,上述结果需要去除重复的相同行,并合并不同行中的相同ids。去除重复行得到543 460行,然后每行内的id排序后将每行第一个id相同的行进行合并,得到76 337行,再次进行每行第一个id排序合并后得到39 572行。然后可以将每行内id代表的各个序列下一步用phrap获得重叠群,结果见表2.

表2 Blast比对水稻所有的EST两两序列并合并序列相似性打分达到100以上序列ids
2.4用phrap拼接获得EST重叠群
根据前述方法用phrap程序从前面的blast比对结果进行重叠群拼接,获得只有一个重叠序列的重叠群为27 556个,两个以上超过一个重叠序列的为7 413个,所有重叠群序列总数达到171 698个(见图4)。为了找出更合适的比对重叠群,将获得的重叠群两两进行了blast但打分大于250,这样获得了34 969个比对结果,其中16 900个为单一序列(见图5),这样为下一步进行clustalw比对分析很有帮助(本文未附)。

图4 Blast结果用phrap进行重叠群拼接

图5 Phrap得到的重叠群进行blast比对,显示了3行,每行超过一个重叠群的彼此相似性打分超过250
2.5重叠群与NCBI nt数据库比对进行注释
获取重叠群的注释尤为重要,将重叠群与NCBI nt核酸数据库进行比对后从返回的信息中挖掘各个重叠群的注释。全部的34 969个重叠群与NCBI nt数据库进行blast比对后,1 971个没有返回比对结果,注释内容提取合为一行如图6所示。去除重复行后注释行总数为211 351,但其中还有相当部分为未注释的行,如在含有chromosome, cultivar:, genomic sequence, clone, mRNA sequence, unknown, hypothetical protein, DNA, Cosmid, vector, cDNA, BAC clone, marker等的比对结果中大部分没有有用的注释信息,还需要进一步去除约只有一半为有用的注释行,见图7示例.这些注释内容需要与前面的重叠群进行匹配后进一步进行挖掘。

图6 重叠群与NCBI nt数据库进行blast比对后提取的注释行示例

图7 从注释行中去除非注释行获得的注释行示例,参见上下文分析
物种关联的注释可提供一些有意义的信息,尤其是对于比较基因组学分析。从比对结果中找出了939个物种与水稻重叠群有关联,只有82个物种出现的注释超过100条,而其中仅仅10个超过了1 000条。玉米与水稻的比对注释最多达到了36 804条,大多数为mRNA/cDNA/protein的注释也许可以提供与基因功能相关的有用信息。剩下的9个中只有Brachypodium distachyon超过1万条,为11 610条,见图8。
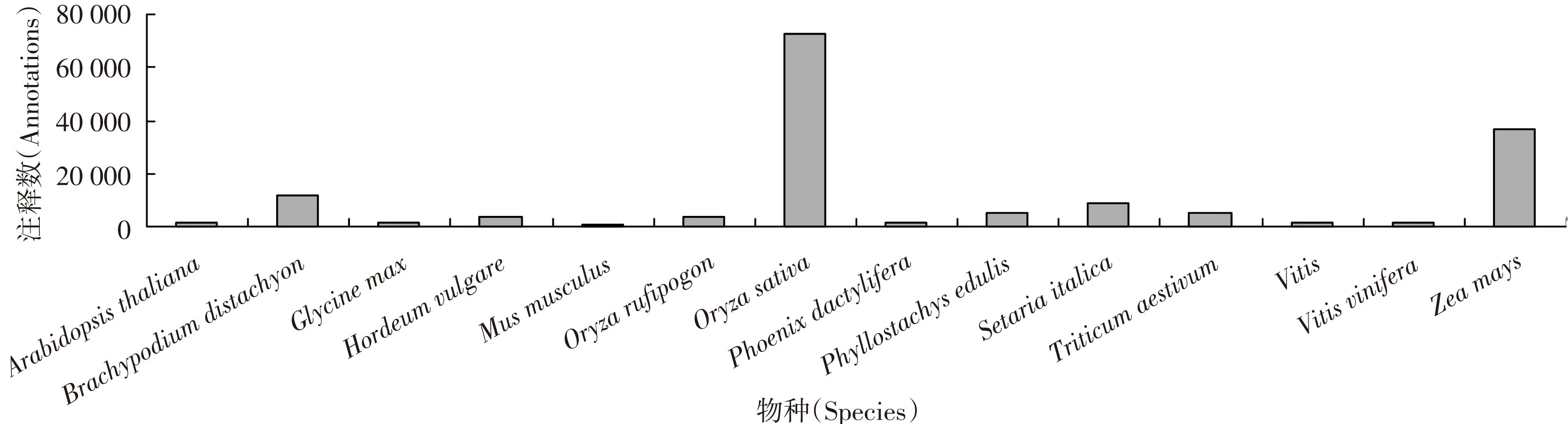
图8 不同物种与水稻重叠群比对返回超过1 000条的注释数
每个重叠群比对结果出现推测基因功能的注释对于进一步的功能基因组学分析特别是构建基因调控网络是很有帮助的,这将是我们下一步的研究目标。
2.6水稻发育过程组织特异表达
确定组织特异转录谱对于分析基因表达模式及构建基因调控网络是很重要的。所有的EST记录中见图9,其中花药的记录数最多。从比对的39 572个EST id(见图2)组找出了各个组织的表达重叠群,结果见图10,虽然表达重叠群中可能含有相似的重叠群,如图5所示。从图10可以清楚看出,花药的表达重叠群最多达到了最高重叠群数。这并奇怪,因为从花药的EST总数977 141(见图9)可以预见(分析的EST序列总数只有125万条),其他的组织都少于20万条。从以上结果尚不能完全的获得组织特异表达谱(见图10),但是很显然组织特异表达谱对于构建水稻发育过程基因调控网络是很重要的,我们将在今后继续进行探讨。

图9 本文引用的NCBI来源水稻不同组织EST序列数

图10 从比对后EST id组(见图2)获取的不同组织表达重叠群计数
3讨论
EST大数据包含了大量基因表达信息,EST数据大规模分析有助于发现基因调控的活动情况,并可以用于构建基因调控网络。本文从NCBI下载了水稻的125万条EST序列并进行了基因表达分析。所有的分析工作都是通过FreeBSD操作系统完成的,主要工具包括Unix命令,及本地blast,phrap及远程blast程序(见方法)。经过blast 比对,phrap重叠群拼接及再比对,获得了34 969重叠群,其中约一半只有一个重叠群序列(见图5)。进一步我们将重叠群序列与NCBI全长cDNA获取的单一基因进行比对以获得水稻的完整转录组。以上结果表明,我们进行的大规模EST分析是有效且快捷,与其他方法相比并不需要复杂的算法[3,10]。
本文初步分析了水稻的组织特异表达谱,发现花药表达的EST重叠群数量最多,其他组织较少些,原因尚未进一步分析(见图10)。通过与NCBI核酸数据库进行远程比对,从返回结果中提取了每个重叠群的注释信息(见图6~图8),今后我们将着重挖掘特异表达基因并进一步构建水稻发育过程的基因调控网络。
参考文献(References)
[1]GIALLOURAKIS C C, BENITA Y, MOLINIE B, et al. Genome-wide analysis of immune system genes by expressed sequence Tag profiling[J]. J Immunol, 2013,190(11):5578-87.
[2]SHA A H, LI C, YAN X H, et al. Large-scale sequencing of normalized full-length cDNA library of soybean seed at different developmental stages and analysis of the gene expression profiles based on ESTs[J]. Mol Biol Rep, 2012,39(3):2867-74.
[3]MENON R, GARG G, GASSER R B, et al. TranSeqAnnotator: large-scale analysis of transcriptomic data[J]. BMC Bioinformatics, 2012,13( Suppl 17):S24.
[4]ZHU W, BUELL C R. Improvement of whole-genome annotation of cereals through comparative analyses[J]. Genome Res, 2007, 17(3):299-310.
[5]WARD J A, PONNALA L, WEBER C A. Strategies for transcriptome analysis in nonmodel plants[J]. Am J Bot, 2012, 99(2):267-76.
[6]LUO H, SUN C, LI Y, et al. Analysis of expressed sequence tags from the Huperzia serrata leaf for gene discovery in the areas of secondary metabolite biosynthesis and development regulation[J]. Physiol Plant, 2010, 139(1):1-12.
[7]FRAZIER T P, ZHANG B. Identification of plant microRNAs using expressed sequence tag analysis[J]. Methods Mol Biol, 2011, 678:13-25.
[8]VICTORIA F C, DA MAIA L C, DE OLIVEIRA A C. In silico comparative analysis of SSR markers in plants[J]. BMC Plant Biol, 2011, 11:15.
[9]XIE F, SUN G, STILLER J W, et al. Genome-wide functional analysis of the cotton transcriptome by creating an integrated EST database[J]. PLoS One, 2011, 6(11):e26980.
[10]LI Y, GONG P, PERKINS E J, et al. RefNetBuilder: a platform for construction of integrated reference gene regulatory networks from expressed sequence tags[J]. BMC Bioinformatics, 2011, 12( Suppl 10):S20.
[11]SONG D G, ZHANG H S, HUANG L X, et al. Localization, Updating and Sequence Preprocessing of EST Database under Unix Environment[J]. Chinese Journal of Bioinformatics, 2010,8(1):52-56.
*通信作者:姚玉华,男,教授,研究方向:计算生物学,应用数学;E-mail: yaoyuhua@zstu.edu.cn.
猜你喜欢
幼儿100(2023年39期)2023-10-23 11:36:32
青少年科技博览(中学版)(2022年6期)2022-12-27 19:44:27
中国土壤与肥料(2021年5期)2021-12-12 02:02:11
今日农业(2021年21期)2021-11-26 05:07:00
军事文摘(2021年22期)2021-11-26 00:43:51
今日农业(2021年14期)2021-10-14 08:35:40
金桥(2021年7期)2021-07-22 01:55:38
今日农业(2020年20期)2020-11-26 06:09:10
文苑(2020年6期)2020-06-22 08:41:52
文苑(2019年22期)2019-12-07 05:29:00
