
基于氨基酸约化和统计特征的蛋白质亚细胞定位预测
2016-01-15 02:06:16杨红,徐慧敏,严寿江等
生物信息学 2015年2期
关键词:支持向量机
基于氨基酸约化和统计特征的蛋白质亚细胞定位预测
杨红1,2,徐慧敏2,严寿江2,陈静2,耿丽丽2,姚玉华2*
(1.青岛滨海学院,青岛 266555;
2.浙江理工大学生命科学学院,杭州 310018)
摘要:蛋白质亚细胞定位预测对蛋白质的功能、相互作用及调控机制的研究具有重要意义。本文基于物化性质和结构性质对氨基酸的约化,描述序列局部和全局信息的“组成”、“转换”和“分布”特征,并利用氨基酸亲疏水性的数值统计特征,提出了一种新的蛋白质特征表示方法(NSBH)。分别使用三种分类器KNN、SVM及BP神经网络进行蛋白质亚细胞定位预测,比较了几种方法和特征融合方法的预测结果,显示融合特征表示及结合SVM分类器时能够达到更好的预测准确率。同时,还详细讨论了不同参数对实验结果的影响,具体的实验及比较结果显示了该方法的有效性。
关键词:蛋白质亚细胞定位;氨基酸物化性质;支持向量机
中图分类号:Q811文献标志码:A
收稿日期:2015-03-05;修回日期:2015-04-08.
基金项目:生物信息学数据库技术课程教学改革,福建农林大学研究生教学改革项目(No.112501234)。
作者简介:郑珂晖,男,讲师,博士研究生,研究方向:生物信息学;E-mail:zhkehui@tom.com.
doi:10.3969/j.issn.1672-5565.2015.02.06
Protein subcellular localization prediction based on reduced representation of
amino acid and statistical characteristic
YANG Hong1,2,XU Huimin2,YAN Shoujiang2,CHEN Jing2,GENG Lili2,YAO Yuhua2*
(1.QingdaoBinhaiUniversity,Qingdao266555,China;
2.CollegeofLifeSciences,ZhejiangSci-TechUniversity,Hangzhou310018,China)
Abstract:The protein subcellular localization prediction is important to study the protein function, protein interaction and their regulation mechanism. In this paper, based on four amino acids physicochemical properties and structural properties,We describe the local and global information of sequence by ‘component’, ‘transition’ and ‘distribution’. Using the numerical statistical characteristic of hydrophobic/hydrophilic amino acid, we proposed a new protein feature representation. We compare the prediction results between the proposed methods and fusion method with the classification algorithm KNN, SVM and BP. The results show that fusion method with SVM can get better prediction accuracies. Meantime, we also discuss the effects of different parameters on the experimental results. The detailed experimental and comparison results show the effectiveness of the proposed method.
Keywords:Subcellular localization; Physicochemical properties; Support vector machine (SVM)
蛋白质是生命的物质基础,是构成细胞的基本有机物,是生命活动的主要承担者。然而,蛋白质只有在特定的亚细胞位置中才能行使其特定的功能。随着高通量技术的发展,很大数量的蛋白质序列正日益增长并被整理和存入到公共的生物数据库。根据2014年2月发布的统计表明,UniProtKB/Swiss-Prot包含542 258条序列,然而在1986年仅仅是3 939条[1]。采用实验方法确定蛋白质的亚细胞定位需要耗费大量的人力、物力、财力,已经无法满足数据库中蛋白质序列爆炸性增长的现实需要,从已积累的知识和数据出发,开发蛋白质亚细胞定位预测的计算机方法就成为了当前的重要研究任务。
在使用计算方法来确定蛋白质亚细胞位置的研究方面,人们已经做出了很多的尝试[2-7],近年来这方面已经做的更加完善。在最近几年的文章中,这方面的大部分贡献是由Chou和Shen做出的。其他相关的作者也提供了一些在线的服务平台来帮助解决蛋白质亚细胞定位的相关问题[8-18]。最初Nakashima和Nishikawa提出氨基酸组成信息用于判别细胞内和细胞外的蛋白质[19]。随后基于序列的方法,人们又提出二肽组成,间隔氨基酸对组成和伪氨基酸组成[20-22]。为了研究序列的进化信息,随后人们又提出序列的位置特异性得分矩阵(PSSM)[23-24]。近几年随着基因本体论(GO)数据库的不断更新,人们提出基于基因本体论的计算方法来预测蛋白质亚细胞定位等问题[25-27]。不仅是蛋白质序列信息,分类算法也能够影响蛋白质亚细胞定位预测。到目前为止许多计算技术如隐马氏模型(HMM)[28],神经网络[29],K-近邻(KNN)[27, 30]和支持向量机(SVM)被用于分类预测[31-33]。
本文提出了四种氨基酸物化性质和结构性质并提出新的蛋白质特征表示方法——基于氨基酸亲疏水性的数值统计特征(NSBH)。特征信息的融合与支持向量机的结合达到了较好的实验结果。
1数据和方法
1.1数据集
本文使用了两组数据集,两组数据集在同一个亚细胞位置子集中蛋白质之间的序列一致性≤25%。第一组数据集是NNPSL数据集,这个数据集最先是由Reinhardt和Hubbar建立的[34]。它包含997条原核蛋白,分为三个亚细胞位点和2 427条真核蛋白,分为四个亚细胞位点。数据集里的所有蛋白质都是从SWISS-PROT 33.0中提取的,并且没有跨膜蛋白。在每一个亚细胞位置中,没有一条序列与其他任何一条序列的相似度大于90%。第二组数据集是Cell-PLoc 2.0包其包含下列六个子数据集:Euk-mPLoc、Hum-mPLoc、Plant-mPLoc、Gpos-mPLoc、Gneg-mPLoc和Virus-mPLoc[12]。它们适用于真核、人类、植物、革兰氏阳性菌、革兰氏阴性菌和病毒蛋白质。另外,Cell-PLoc 2.0中基准数据集的构建是基于SWISS-PROT 55.3。数据集涵盖22个亚细胞位点,在同一个亚细胞位置子集中蛋白质序列之间的一致性≤25%。Cell-PLoc 2.0包可以从http://www.csbio.sjtu.edu.cn/bioinf/Cell-PLoc-2/中免费得到。
1.2方法
蛋白质亚细胞定位预测的关键步骤是有效的数学表达式把蛋白质符号特征转换为与预测任务相关的特征向量以及分类算法辨别特征向量。因此,我们将重点介绍这两部分。
1.2.1序列特征信息提取
本文使用了关于氨基酸序列的局部和全局信息。主要包括四种常用的氨基酸物化性质,氨基酸组分信息(Amino acid composition)以及我们提出的一种基于氨基酸亲疏水性的数值统计特征(NSBH)。
(1)常用的氨基酸物化性质和结构性质
在文中,我们用到了四种常见的氨基酸物化性质和结构性质分别为疏水性(Hydrophobicity)、归一化范德华体积(Normalized van der Waals volume)、极性(Polarity)和极化性(Polarizability)。
一条蛋白质序列由基于不同物化性质和结构性质的参数向量所表示,这些参数向量包含“组成”(Composition),“转换”(Transition)和“分布”(Distribution)三种描述符[35],具体见表1。他们分别用来描述一条蛋白质中一个给定氨基酸性质的全局组成,沿着整条蛋白质的性质改变的频率,以及沿着序列的性质的模式分布。
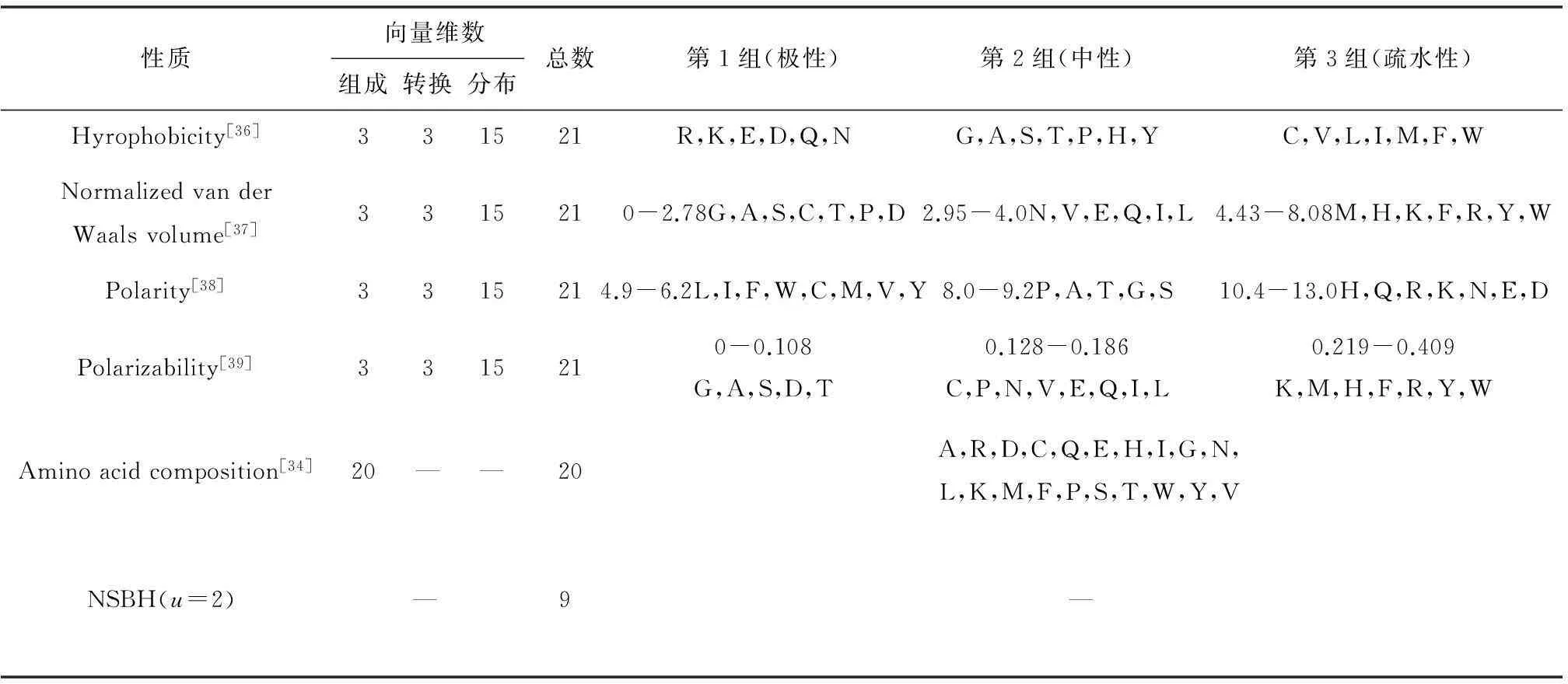
表1 氨基酸属性和每一种性质的特征维数
(2)氨基酸组份
氨基酸组份与蛋白质亚细胞定位有一定的关联。不同类型的蛋白质通常需要不同的氨基酸组成对应于特定的生理功能。因此,对于细胞核的定位,组氨酸的丰富含量可以看作是一个特征。
给定一条蛋白质序列P,序列可以表示为
(1)
其中,fi(i=1,2,…,20)是蛋白质P中20种氨基酸的归一化的发生频率,T是转置运算符。许多预测蛋白质亚细胞定位的方法是基于氨基酸组份信息。
(3)NSBH特征方法
这里我们考虑一个与蛋白质结构有重要关系的物化性质:氨基酸的疏水性。首先,每一个氨基酸被它自己的物化性质所特征化。20种氨基酸被约化为两种类型:疏水氨基酸H={F,L,I,Y,M,W,V,A,P,C};亲水氨基酸:P={S,N,K,D,R,T,H,Q,E,G}。然后20种氨基酸进一步约化为四种类型:强疏水氨基酸:SH={F,L,I,Y,W};弱疏水氨基酸:WH={M,V,A,P,C};强亲水氨基酸:SP={S,N,K,D,R};弱亲水氨基酸:WP={T,H,Q,E,G}。
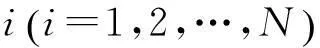
(2)
对于一条数值序列,我们计算u个连续数值的和,这些和被看作振幅。为了得到蛋白质序列的数值表示,我们计算振幅的频率。因此,一条蛋白质序列可以被一个向量特征化。例如当u=2时,振幅是-4,-3,-2,-1,0,1,2,3,4。通过计算振幅的频率,一条蛋白质序列可以被一个9维的向量特征化。
最后,基于以上六种特征信息,我们构建其融合模型。在融合模型中,每一条蛋白序列被表示为113维的向量。
1.2.2预测方案
本文使用了三种分类器:支持向量机(SVM),K近邻(KNN),BP神经网络。下面将一一详细介绍。
(1)本文首先采用了Vapnik的支持向量机来预测亚细胞定位[40]。然而蛋白质亚细胞定位预测是一个多分类问题。因此,我们采用了多类预测方法。支持向量机采用“one-versus-rest”策略,给定一条未知测试蛋白序列,提取其特征向量之后输入SVM。SVM首先把输入向量映射到一个特征空间,然后SVM寻找一个最优线性决策来解决特征空间中两类或多类问题,最后,一个预测标签被分配给测试蛋白。在我们的研究中,我们使用LIBSVM来实行SVM分类,选择径向基函数(RBF)作为核函数。
对于SVM,我们选择径向基函数作为核函数是因为相比于其他核函数它优越于解决非线性问题[41]。这里,为了尽可能的得到最高的预测准确率,我们选择了参数。对于每一个数据集,基于10倍交叉验证,我们使用网格搜索策略选择参数c和g的值。则c与g值的范围是2-5到25。
(2)K近邻(K-Nearest Neighbor,KNN)分类算法是一个比较成熟的算法,该算法的方法原理也十分简单。KNN分类算法的思路是:假如一个样本在特征空间中的K个最相似的样本中的大多数属于同一个类别,那么这个样本也属于此类别。这里我们将使用的KNN准则用于分类预测[42-43]。
(3)目前,研究人员构建了许多不同的神经网络模型,本文应用的是反向传播神经网络(Back Propagation Neural Network,即BP神经网络)[29]。在人工神经网络之中,反向传播神经网络是一种稳定性和鲁棒性较强的人工神经网络,另外。它也属于有监督学习的网络模型。本文构建的BP网络,隐层节点参数值为9,隐层和输出层皆采用Sigmoid传输函数,输出维数由各数据集所含亚细胞位点数决定,其它参数采用默认值。
1.2.3评价方法
本文中,我们使用留一法验证来评估我们方法的预测结果。留一法是指仅选择原样本中的一项作为测试样本,而剩余的留作训练样本。这个样本一直持续到每个样本都被作为一次测试样本。我们使用Overall accuracy作为本文的预测结果评价指标:
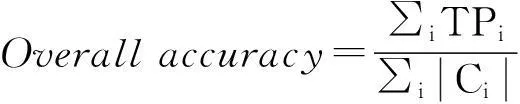
(3)
2结果与分析
2.1NSBH方法中不同u值结果比较
从图1中可以看出,当u从1变化到10时,除了病毒数据集的预测结果准确率变化幅度大一点外,其他数据集的结果都处于一个平缓状态。RH997,RH2427,革兰氏阳性菌和革兰氏阴性菌的结果相对较好;病毒,人类,真核和植物的预测结果相对就差一点。
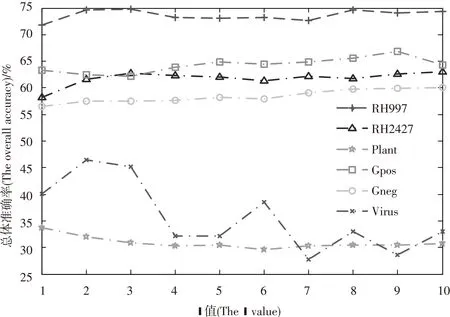
图1 每个数据集上不同 u值SVM分类器结果比较
从图1中明显看出,随着u值在1~10范围内的变化,除了病毒数据集外其他各数据集的总体准确率呈平稳状态。病毒数据集随着u值的变化呈现波浪式变化,但总的趋势还是随着u值的增大而下降。可能是由于较其他数据集病毒数据集数据个数较少,而蛋白质位点个数相对偏多,从而导致预测结果不是很稳定。考虑到病毒数据集在u=2时准确率较其他k值要高出许多,且向量的维数偏低,降低实验运行的时间,因此,我们可认为u=2时总体达到较好的结果。此时,病毒数据集的准确率为46.43%。
2.2不同特征信息提取方法的比较
表2列出了八个数据集中不同的特征信息提取方法的结果比较。基于上述分析,在NSBH方法中,当u=2时总体预测结果较好,所以在此部分只需考虑u=2时与其他方法的比较。从表中可以明显看出融合后的信息准确率明显高于单个性质的预测准确率,该现象可能是由于融合信息包比单个特征信息含了更多蛋白质序列的核心特征。
从总体数据集的单个特征信息预测结果来看,AAC结果相对较好,其次是疏水性、极性。而极化性和归一化范德华体积的预测结果就相对差点。本文新提出的方法NSBH,其预测结果相比于传统的氨基酸组成来说不是很理想,但这也是蛋白质序列特征表示的一种新颖的方法。
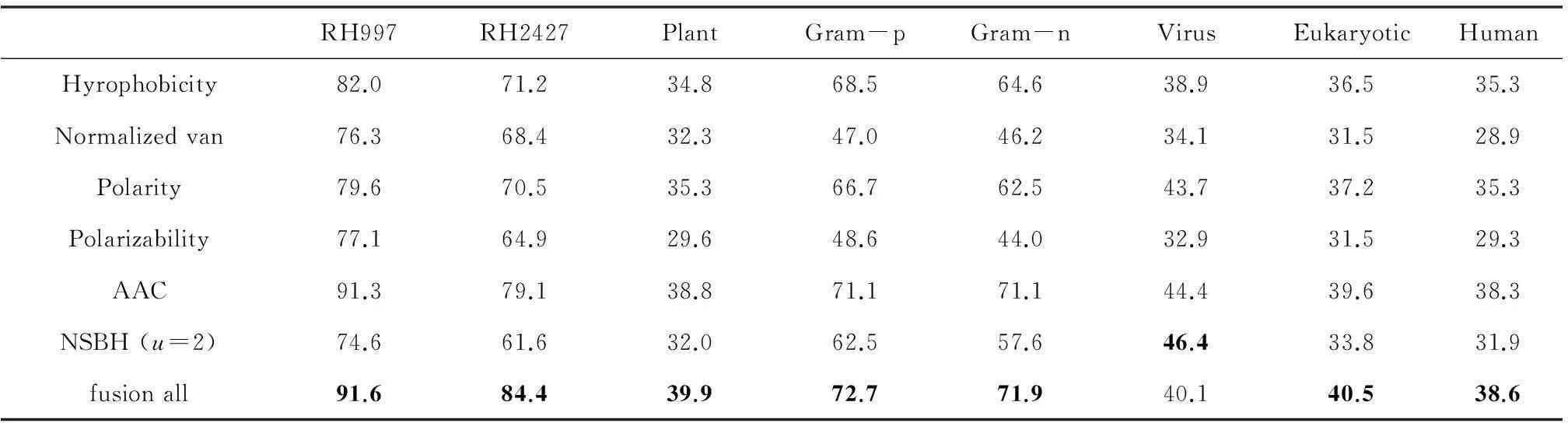
表2 每一个数据集中不同特征提取方法SVM分类器结果比较(%)
2.3KNN分类器中不同k值结果比较
对两组数据集中的每一条序列进行本文使用的5种氨基酸的物化性质信息以及氨基酸组份信息进行融合,最后得到113维的向量。由表5分析得到,融合后的特征表示比单个特征表示预测结果要好,在此,我们分析了基于融合特征表示的不同类型的KNN分类器对预测结果的影响如图2所示。
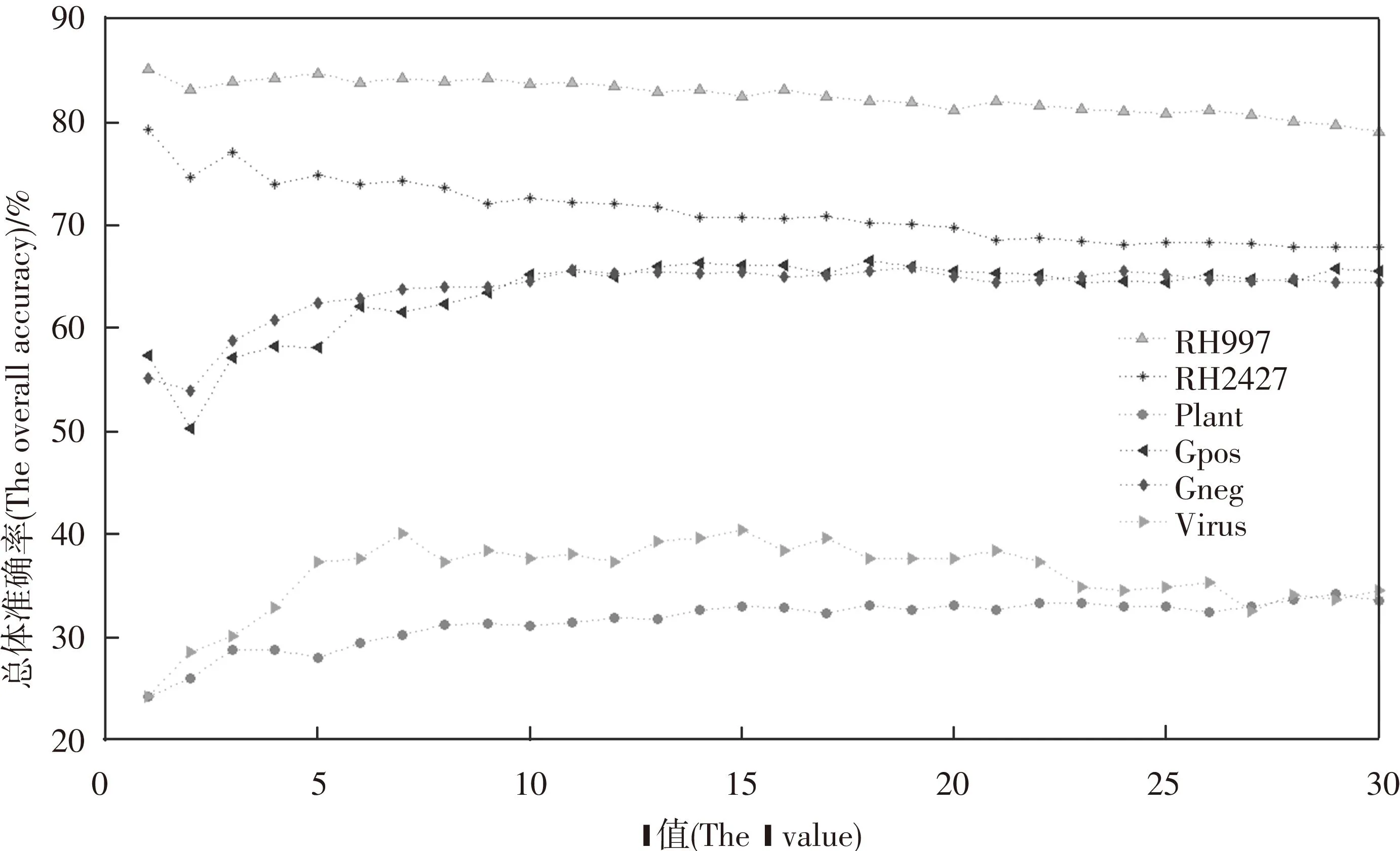
图2 基于融合特征表示的不同类型的KNN分类器比较
从图2可以得出与图1相一致的结论:RH997,RH2427,革兰氏阳性菌和革兰氏阴性菌的结果相对较好;病毒,人类,真核和植物的预测结果相对差一点。对于RH997和RH2427,随着k值的增加,预测结果有下降的趋势并趋于平缓。对于其他六个数据集的结果,随着k值的增加,预测结果有上升的趋势并趋于平缓。对于每个数据集,从整体预测结果及实验运行耗时来看,当k=7时总体预测结果较好。
2.4不同分类器预测结果比较
表3给出的是每个数据集在相应的k值下取得的最好预测结果与SVM预测结果的比较。括号中的k值是相应的数据集在KNN分类器中取得最好预测结果时的值。

表3 基于融合特征下的KNN, SVM和BP预测结果比较
从表3可以明显得出,对于病毒数据集,BP的预测结果是最高的,KNN的最好预测结果稍微高于SVM预测结果。而其他数据集中,SVM的预测结果都比KNN中的最好预测结果和BP的预测结果要高,高出值范围分别为5~6.4个百分点和4.2~9.9个百分点。因此,总体来说,SVM分类器的表现更加的优异。
2.5结果比较分析
为了论证本文提出的方法的可靠性,对于RH997和RH2427这两个数据集,我们的方法与其他方法结果进行了比较。这里给出的是SVM预测结果与其他方法通过留一法检验对NNPSL数据集的预测结果比较。
从表格4中明显看出本文方法的预测总体准确率除了比Chou and Cai的结果低3.1%和比Niu等人略低外,比其他方法高了2.5~5.1个百分点。
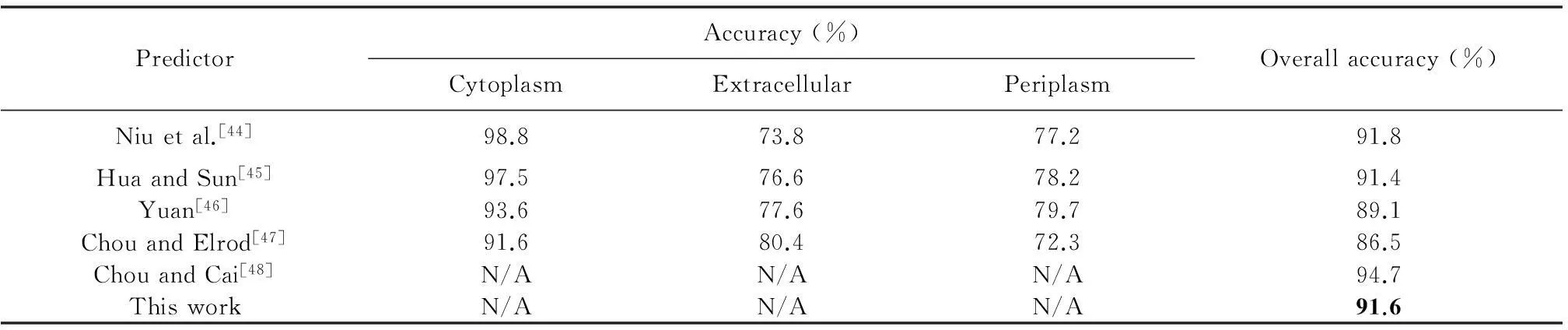
表4 RH997上不同预测模型的预测结果
同样的,从表格5中可以得到除了Chou and Cai方法的结果高于本文结果外,其他研究者方法的结果均低于本文方法结果的3.6~11.4个百分点。
以上两个表格的结果比较得出基于本文提出的新方法NSBH与其他方法的融合再结合常用的SVM分类器能够得到较好的分类效果。因此,本文的方法也可用于蛋白质等其他方面的应用。
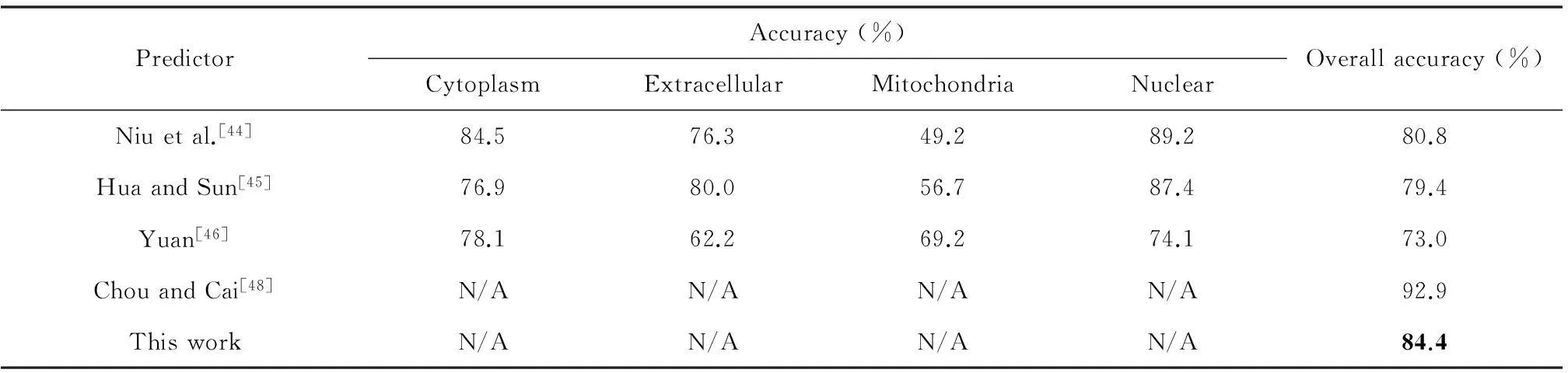
表5 RH2427上不同预测模型的预测结果
3讨论
到目前为止,有许多信息提取算法都是基于氨基酸残基的物化性质,然而单纯的使用这些物化性质进行序列的特征信息提取难免会丢失许多序列的核心特征,所以通常情况下我们都是结合其他特征一起使用。本文尝试使用常见氨基酸的物化性质和结构性质的结合进行蛋白质亚细胞定位预测研究。当然,本文提出的方法也可以应用到其他方面,比如蛋白质结构类预测,蛋白质功能预测方面等。
在K近邻算法中,关键问题主要有确定距离函数和决定k的取值。然而距离函数的确定比较困难且分类的结果与参数有关,在进行训练时,K近邻还需要大量的训练数据,这些都导致K近邻算法在实际应用中存在许多问题。BP神经网络可变参数太多,训练时间需求很大,对固定训练样本的过度拟合会使得预测性能降低。而支持向量机属于一般化线性分类器,SVM的特点是可以同时最小化经验误差和最大化几何边缘。因此与K近邻和BP神经网络相比,SVM分类器更能达到精确值。
参考文献(References)
[1]LI L, YU S, XIAO W, et al. Prediction of bacterial protein subcellular localization by incorporating various features into Chou′s PseAAC and a backward feature selection approach[J]. Biochimie, 2014, 104: 100-107.
[2]CHOU G, WU Z, XIAO X i.Loc-Euk: a multi-label classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins[J]. PLoS One, 2011, 6(3):e18258.
[3]LI L, ZHANG Y, ZOU L, et al. An ensemble classifier for eukaryotic protein subcellular location prediction using gene ontology categories and amino acid hydrophobicity[J]. PLoS One, 2012, 7(1): e31057.
[4]CAI Y, HE J, LI X, et al. Prediction of Protein Subcellular Locations with Feature Seclection and Analysis[J]. Protein &Peptide Letters, 2010, 17(4): 464-472.
[5]WAN S, MAK M, KUNG S. HybridGO-Loc: mining hybrid features on gene ontology for predicting subcellular localization of multi-location proteins[J]. PLoS One, 2014, 9(3): e89545.
[6]MEI S. Predicting plant protein subcellular multi-localization by Chou′s PseAAC formulation based multi-label homolog knowledge transfer learning[J]. Journal of Theoretical Biology, 2012, 310: 80-87.
[7]DEHZANQI A, HEFFERNAN R, SHARMA A, et al. Gram-positive and Gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chou's general PseAAC[J]. Journal of Theoretical Biology, 2015, 364: 284-294.
[8]EMANUELSSON O, NIELSEN H, BRUNAK S. Predicting sub-cellular localization of proteins based on their N-terminal amino acid sequence[J]. Journal of Molecular Biology, 2000, 300(4): 1016.
[9]CHOU K, CAI Y. A new hybrid approach to predict subcellular localization of proteins by incorporating gene ontology[J]. Biochemical and Biophysical Research Communications, 2003, 311: 743-747.
[10]YU C, LIN C, HWANG J. Predicting subcellular localization of protein for Gram-negative bacteria by support vector machines based on n-peptide compositions[J]. Protein Science, 2004, 13(5): 1402-1406.
[11]HORTON P, OBAYASHI T. WoLF PSORT:protein subcellulur localization predictor[J]. Nucleic Acids Research, 2007, 35: W587.
[12]CHOU K, SHEN H. Cell-PLoc 2.0: An improved package of web-servers for predicting subcellular localization of proteins in various organisms[J]. Natural Science, 2010, 2: 1090.
[13]BRIESEMEISTER S, RAHNENFUHRER J, KOHLBACHER O. Going from where to why-interpretable prediction of protein subcellular localization[J]. Bioinformatics, 2010, 26(9): 1232-1238.
[14]YUN N, WAQNER J, LAIRD M, et al. PSORTB 3.0: improved protein subcellular localization prediction with refined localization sub-categories and predictive capabilities for all prokaryotes[J]. Bioinformatics, 2010, 26(13): 1608-1615.
[15]PIERIEONI A, MARTELLI P, CASADIO R. MemLoci: predicting subcellular localization of membrance proteins in eukaryotes[J]. Bioinformatics, 2011, 27(9): 1224-1230.
[16]WAN S, MAK M, KUNG S. mGOASVM:multi-label protein subcellular localization based on gene ontology and support vector machines[J]. BMC Bioinformatics, 2012, 13(11): 290.
[17]CHANG T, WU L, LEE T, et al. EuLoc: a Web-server for accurately predict protein subcellular localization in enkaryotes by incorporating various features of sequence segments into the general form of Chou's PseAAC[J]. Journal of Computer-Aided Molecular Design, 2013, 27(1): 91-103.
[18]WANG X, LI G, LU W. Virus-ECC-mPLoc: a multi-label predictor for predicting the subcellular localization of virus proteins with both single and multiple sites based on a general form of Chou′s pseudo amino acid composition [J]. Protein and Peptide Letters, 2013, 20(3): 309-317.
[19]NAKASHIMA H, NISHIKAWA K. Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies[J]. Journal of Molecular Biology, 1994, 238: 54-61.
[20]ZUO Y, PENG Y, LIU L, et al. Predicting peroxidase subcellular location by hybridizing different descriptors of Chou’ pseudo amino acid patterns[J]. Analytical Biochemistry, 2014, 458: 14-19.
[21]DU P, GU S, JIAO Y. PseAAC-General: Fast Building Various Modes of GeneralForm of Chou’s Pseudo-Amino Acid Composition forLarge-Scale Protein Datasets[J]. International Journal of Molecular Sciences, 2014, 15: 3495-3506.
[22]MANDAL M, MUKHOPADHYAY A, MAULIK U. Prediction of protein subcellular localization by incorporating multiobjective PSO-based feature subset selection into the general form of Chou’s PseAAC[J]. Medical & Biological Engineering & Computing, 2015, 53: 331-344.
[23]JEONG J, LIN X, CHEN X. On Position-Specific Scoring Matrix for Protein Function Prediction[J]. IEEE Transactions on Computational Biology and Bioinformatics, 2011, 8: 308-315.
[24]HUANG C, YUAN J. Using radial basis function on the general form of Chou’s pseudoamino acid composition and PSSM to predict subcellular locations ofproteins with both single and multiple sites[J]. BioSystems, 2013, 113: 50-57.
[25]WAN S, MAK M, KUNG S. GOASVM: A subcellular location predictor by incorporating term-frequencygene ontology into the general form of Chou’s pseudo-aminoacid composition[J]. Journal of Theoretical Biology, 2013, 323: 40-48.
[26]MAZANDU G, MULDER N. The use of semantic similarity measures for optimally integrating heterogeneous Gene Ontology data from large scale annotation pipelines[J]. Frontiers in Genetics, 2014, 5: 264.
[27]XIAO X, WU Z, CHOU K. iLoc-Virus: A multi-label learning classifier for identifying the subcellular localization of virus proteins with both single and multiple sites[J]. Journal of Theoretical Biology, 2011, 284: 42-51.
[28]LIN T, MURPHY R, BAR-JOSEPH Z. Discriminative motif finding forpredicting protein subcellular localization[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2011, 8(2): 441-451.
[29]ZOU L, WANG Z, HUANG J. Prediction of subcellular localization ofeukaryotic proteins using position-specific profiles and neural network withweighted inputs[J]. Journal of Genetics and Genomics, 2007, 34(12): 1080-1087.
[30]XIAO X, WU Z, CHOU K. A Multi-Label Classifier for Predicting the Subcellular Localization of Gram-Negative Bacterial Proteins with Both Single and Multiple Sites[J]. PLoS One, 2011, 6: e20592.
[31]CAI Y, LIU X, XU X, et al. Support vector machines for prediction of protein subcellular location by incorporating quasi-sequence-order effect[J]. Journal of Cellular Biochemistry, 2002, 84: 343-348.
[32]LIANG R, HUANG S, SHI S, et al. A novel algorithm combining support vector machine with the discretewavelet transform for the predict ion of protein subcellular localization[J]. Computers in Biology and Medicine, 2012, 42: 180-187.
[33]LIU T, TAO P, LI X, et al. Prediction of subcellular location of apoptosis proteins combing trigram encoding based on PSSM and recursive feature elimination[J]. Journal of Theoretical Biology, 2015, 366: 8-12.
[34]REINHARDT A, HUBBARD T. Using neural networks for prediction of the subcellular location of proteins[J]. Nucleic Acids Research, 1998, 26: 2230-2236.
[35]DUBCHAK I, MUCHNIK I, HOLBROOK S, et al. Prediction of protein folding class using global description of amino acid sequence[J]. Proceedings of the National Academy of Sciences, 1995, 92: 8700-8704.
[36]CHOTHIA C, FINKELSTEIN A. The classification and origins of protein folding patterns[J]. Annual Review of Biochemistry, 1990, 59: 1007-1035.
[37]FAUCHERE J, CHARTON M, KIER L, et al. Amino acid side chain parameters for correlation studies in biology and pharmacology[J]. International Journal of Peptide and Protein Research, 1988, 32: 269-278.
[38]GRANTHAM R. Amino acid difference formula to help explain protein evolution[J]. Science, 1974, 185: 862-864.
[39]CHARTON M, CHARTON B. The structural dependence of amino acid hydrophobicity parameters[J]. Journal of Theoretical Biology, 1982, 99: 629-644.
[40]CORTES C, VAPNIK V. Support-Vector Networks[J]. Machine Learning, 1995, 20(3):273-297.
[41]YUAN Z. Better prediction of protein contact number using a support vector regression analysis of amino acid sequence[J]. BMC Bioinformatics, 2005, 6: 248.
[42]COVER T, HART P. Nearest neighbour pattern classification[J]. IEEE Transactionon Information Theory, 1967, 13: 21-27.
[43]DENOEUX T. A k-nearest neighbor classification rule based onDempster-Shafer theory[J]. IEEE Transactions on Systems Man and Cybernetics, 1995, 25: 804-813.
[44]NIU N, JIN Y, FENG K, et al. Using AdaBoost for the predicting of subcellular location of prokaryotic and eukaryotic proteins[J]. Molecular Diversity, 2008, 12: 41-45.
[45]HUA S, SUN Z. Support vector machine approach for protein subcellularlocalization prediction[J]. Bioinformatics, 2001, 17: 721-728.
[46]YUAN Z. Prediction of protein subcellular locations using Markov chainmodels[J]. FEBS Letters, 1999, 451: 23-26.
[47]CHOU K, ELROD D. Using discriminant function for prediction of subcellular location of prokaryotic proteins[J]. Biochemistry and Biological Physics Research Communications, 1998, 252: 63-68.
[48]CHOU K, CAI Y. A new hybrid approach to predict subcellular localizationof proteins by incorporating gene ontology[J]. Biochemistry and Biological Physics Research Communications, 2003, 311: 743-747.
*通信作者:温永仙,女,教授,博士生导师,研究方向:生物信息学;E-mail:wen9681@sina.com.
猜你喜欢
现代电子技术(2016年23期)2017-01-12 09:57:47
现代电子技术(2016年23期)2017-01-12 09:26:09
无线互联科技(2016年13期)2017-01-10 02:49:43
中国水运(2016年11期)2017-01-04 12:26:47
软件导刊(2016年11期)2016-12-22 21:52:38
电子技术与软件工程(2016年20期)2016-12-21 10:21:33
价值工程(2016年32期)2016-12-20 20:36:43
价值工程(2016年29期)2016-11-14 00:13:35
科学与财富(2016年28期)2016-10-14 21:19:17
考试周刊(2016年53期)2016-07-15 09:08:21
