
萤光素类似物激发态的TD-DFT计算精度评估
2016-01-14 03:27石瑞婷,王炳强,张彩云等
化学研究 2015年5期
关键词:吸收光谱
萤光素类似物激发态的TD-DFT计算精度评估
石瑞婷,王炳强*,张彩云,张坚
(山西师范大学 化学与材料科学学院,山西 临汾 041000)
摘要:采用含时密度泛函理论(TD-DFT)对8种已知萤光素类似物的垂直激发能和发射能进行了系统的考察. 选取10种交换-相关(XC)泛函对8种萤光素类似物的基态和第一单重激发态结构、吸收和发射光谱进行了计算,并将得到的结果与实验数据进行对照. 结果表明,该系列物质吸收光谱和发射光谱的计算对XC泛函的选择非常敏感. B3LYP、mPW3PBE、B3PW91方法能够提供较好理论计算结果,吸收光谱的均方根误差(RMS)在0.40 eV以内,标准差(SD)在0.27 eV以内;发射光谱的RMS在0.24 eV以内,SD在0.17 eV以内.
关键词:萤光素类似物;含时密度泛函;吸收光谱;发射光谱
中图分类号:O641 文献标志码:A
收稿日期:2015-05-06.
作者简介:石瑞婷(1987-),女,硕士生,主要从事量子化学计算研究. *通讯联系人,E-mail:wangbq2007@163.com.
Assessment of TD-DFT in calculations of excited-states
of luciferin analogs
SHI Ruiting, WANG Bingqiang*, ZHANG Caiyun, ZHANG Jian
(SchoolofChemistryandMaterialsScience,ShanxiNormalUniversity,Linfen041000,Shanxi,China)
Abstract:The absorption and emission spectra of eight known luciferin analogs were investigated by time dependent density functional theory (TD-DFT). Geometries and transition energies of eight luciferin analogs were calculated by ten XC functionals and the results were compared with the data of experiment. The results showed the calculations of spectra of luciferin analogs are sensitive to the choice of XC functional. Among the 10 tested functionals, B3LYP, mPW3PBE and B3PW91 yield better theoretical calculation results for absorption and emission spectra. For absorption spectra, the RMS was less than 0.40 eV and SD was less than 0.27 eV; for emission spectra, the RMS was less than 0.24 eV and SD was less than 0.17 eV.
Keywords:luciferin analogs; time-dependent-density functional theory(TD-DFT); absorption spectra; emission spectra
在自然界中许多生物体都具有独特的发光性能,其中人们研究最多的是水母和萤火虫. 自然界中萤火虫的发光效率可达到41.0%±7.4%[1],比一般的电光源物质发光效率大得多(最大效率仅有6%)[2]. 萤火虫体内萤光素[(S)-2-(6′-羟基-2′-苯并噻唑基)-2-噻唑啉-4-羧酸(D-luciferin)]在萤光素酶的催化作用下可以发出多种色彩的可见光,显示出优异的发光性能,引起科学家的密切关注[3-4]. 对于萤光素分子的发光机理研究者们提出了多种假设[3, 5-16],目前最广为人们接受的一种化学原理可分为两大步:(S)-2-(6-羟基-2-苯并噻唑基)-2-噻唑啉-4-羧基(D-LH2)和腺苷-5-三磷酸盐(ATP)生成萤光素-腺苷酸(LH2-AMP);萤光素-腺苷酸被氧化生成氧化萤光素(OXYLH2)、二氧化碳和腺苷-5-磷酸(AMP)[17-20]. 由于萤光素类似物具有发光强度大、发光效率高、发热量低等特点,因而被广泛地应用在医药、ATP的分析测定、微生物检测、生物传感器、生物成像、基因检测报告等方面[1, 21-23].
萤光素类似物的发光过程是分子由能量弛豫的第一激发单重态(S1)向基态(S0)的单电子垂直跃迁,发光颜色由它的发射能(即发射波长)决定,所以对萤光素类似物激发态的准确理论预测将有助于新型荧光有机物的设计、合成和应用. 由于更加精确的理论计算方法如CASPT2、SACCI等在计算生物萤光素方面耗时巨大,而含时密度泛函理论(TD-DFT)可有效地用于中等尺寸或更大体系的激发态研究,因此成为近年来被广泛使用的激发态研究方法之一[1, 24-28]. 然而近期研究表明[24],交换相关(XC)泛函的选择对激发能的计算十分敏感. 评估XC泛函在萤光素类似物激发能计算中的精度是使用TD-DFT方法研究该类化合物发光性质的前提和基础. 本文作者选取8种已知萤光素类似物作为考察对象(图1),其中包括4种酶促反应的反应物,含萘基的氨基取代萤光素 [naphtyl-aminoLH2(NAL)],含喹啉基氨基取代萤光素 [quinolylAL (QAL)],氨基取代萤光素 [aminoLH2(AL)],含香豆素的氨基取代萤光素 [coumarylAL (CAL)];其次还包括4种酶促反应的生成物,即氧化萤光素类似物,氧化萤光素[oxyluciferin (OXYLH2)],脱氧氧化萤光素[dehydroxyOxyLH2(DHOXYLH2)],5,5-二甲基取代氧化萤光素[5,5-dimethyloxyLH2(DMOXYLH2)],6′-甲氧基-5,5-二甲基取代氧化萤光素[6′-methoxy-5,5-dimethyloxyLH2(MDMOXYLH2)]. 我们使用10种XC泛函计算其吸收和发射光谱,并将结果与对应的实验值进行比较、分析,评估泛函的选择对萤光素类似物吸收和发射光谱计算精度的影响. 表1列出了8种萤光素类似物在二甲亚砜(DMSO)溶液中的吸收和发射光谱的实验数据[29].

图1 8种萤光素类似物的化学结构示意图 Fig.1 Chemical structures of eight analogs of luciferin

CompoundALCALNALQALOXYLH2DHOXYLH2DMOXYLH2MDMOXYLH2λab3.292.993.633.263.293.382.143.26λem2.552.632.842.822.772.292.382.48
1计算方法
采用TD-DFT理论对萤光素类似物的激发态进行计算研究,评估了XC泛函对计算精度的影响. 我们评估了8种常见的XC泛函,分别包括:i)1种广义梯度近似泛函(GGA):BLYP;5种杂化的广义梯度近似泛函(H-GGA):B3LYP[30-31],PBE0[32],mPW3PBE[32-33],B3PW91[30],B3P86[30, 34];ii)1种含动能密度的广义梯度近似泛函(M-GGA):BB95[30, 35];iii)1种含动能密度的杂化广义梯度近似泛函(HM-GGA):B1B95[30, 35]. 此外,为了考虑长程校正对激发态计算的影响,选取了2种包含长程校正的XC泛函:CAM-B3LYP[36],ωB97XD[37].
为检验基组对萤光素类似物光谱性质计算的影响,文中采用B3LYP方法和4种不同的基组(6-31+G(d)、6-31++G(d,p)、6-311+G(d)、6-311++G(d,p))对QAL的S0、S1态几何全优化,在相应水平上获得垂直激发能、荧光发射能. 通过对计算结果的比较及对计算精度和计算效率的综合考虑,最终选取6-31+G(d)基组做为后续优化构型和激发态计算使用的基组. 为考察XC泛函对萤光素类似物光谱性质计算精度的影响,分别用10种泛函对上文所述的8种萤光素类似物进行几何全优化计算,并在优化结构的基础上计算了8种物质的垂直激发和发射能. 在相同水平上计算了8种物质基态、激发态的谐振动频率,确保优化所得到的结构确实是势能面上的极小值点. 最后,将计算所得吸收、发射能与实验测得吸收、发射光谱进行比较,并作统计分析. 统计分析包括平均正负误差(mean signed error,MSE),平均绝对误差(mean absolute error,MAE),均方根误差(root-mean-square deviation,RMS),标准差(standard deviation,SD)4个方面[38].MSE用于比较理论计算值相对实验值的高低,若为正值,表明泛函所提供的计算值相较于实验值偏低,反之则偏高;MAE反映理论计算值与实验值的偏离程度;RMS反映理论计算结果相对实验值的精密度;SD反映计算值本身的离散程度[38].
此外,本文作者在B3LYP/6-31+G(d)计算水平下分析了8种萤光素类似物前线分子轨道和自然键轨道(NBO)电荷[39],比较了萤光素类似物从基态跃迁到激发态时电荷转移情况. 以上所有计算均使用PCM模型考虑溶剂化效应,选择DMSO为溶剂. 所有计算项目均采用Gaussian 09程序完成[40].
2结果与讨论
2.1 基组的选择
采用B3LYP与4种不同基组对QAL的基态S0及第一单重激发态S1几何构型、垂直激发能和发射能进行计算(图2和表2).

表2 在B3LYP 计算水平下计算QAL的结构数据、吸收光谱、发射光谱及相应的振子强度(键长单位:nm,光谱能量单位:eV)
图2 QAL的化学结构示意图 Fig.2 Chemical structure of QAL
对于QAL分子,不同基组计算预测的分子结构变化不大. 从6-31+G(d)到6-311++G(d,p),基态键长变化不超过0.000 5 nm,激发态键长变化不超过0.000 7 nm,且均为平面结构. 由表2可见,从6-31+G(d)到6-311++G(d,p),垂直吸收能计算值减小了0.01 eV;垂直发射能增加了0.01 eV. 可见,基组从6-31+G(d)增加到6-311++G(d,p)对QAL基态和激发态结构、能量的计算精度的影响很小,可以忽略不计. 与吸收光谱实验数据3.26 eV对比,计算值偏离了0.1 eV左右;与发射光谱实验数据2.82 eV对比,计算值偏离了不到0.01 eV. 由此可见,不同基组计算所得理论值与实验光谱数据都十分吻合,所选基组均能很好地反映真实实验情况. 考虑计算消耗,在后续所有讨论中,我们将选择6-31+G(d)基组进行计算.
2.2 交换-相关泛函对激发能计算精度的影响
用10种不同DFT泛函对8种萤光素类似物的垂直激发能和发射能进行计算,结果分别列于表3和表4中.
由表3可见,对于垂直激发能,B3LYP、mPW3PBE、B3PW91 3种泛函给出的MSE绝对值最小,|MSE|≤0.11 eV,且均为负值,说明此3种泛函吸收能预测的跃迁能略高于实验值;偏差最大的为ωB97XD、CAM-B3LYP,|MSE|≥0.65 eV. B3LYP、mPW3PBE、B3PW91的MAE最小,均为0.26 eV;ωB97XD、CAM-B3LYP的MAE最大,MAE≥0.67eV,表明这两种泛函计算预测值与实验值偏离较远,误差最大. B3LYP、mPW3PBE和B3PW91泛函的RMS最小,RMS≤0.40 eV,表明这3种泛函对物质光谱性质的预测数据精密度较好,能真实地反映物质的性能. ωB97XD、CAM-B3LYP的RMS值为0.79 eV,是所选泛函中最大的. B3LYP、mPW3PBE和B3PW91 3种泛函的SD值相对较小,SD≤0.27 eV,说明这3种泛函计算值本身的离散程度较小,预测不同萤光类似物的激发能泛函本身引起的误差较小,所得实验数据集中,可信程度高. ωB97XD、CAM-B3LYP的SD值偏大,SD≥0.30 eV,说明这两种长程校正的泛函对萤光类似物激发能的预测数据分散程度比其他泛函预测的数据大.
关于发射能计算的统计数据分析列于表4中.

表3 8种萤光素类似物吸收光谱的 MSE, MAE, RMS
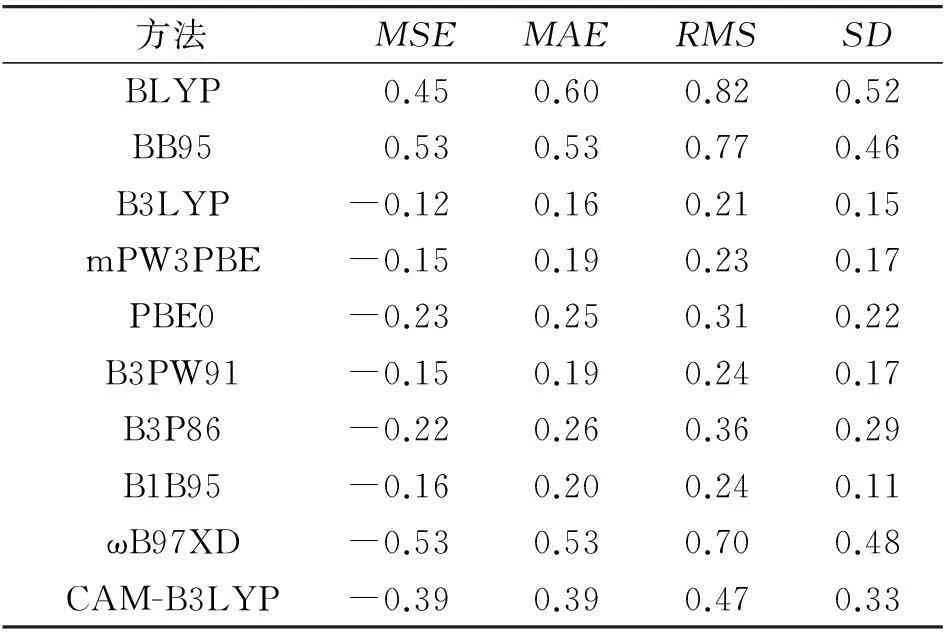
表4 8种萤光素类似物发射光谱的
与垂直激发能计算相似,B3LYP、mPW3PBE、B3PW91 3种泛函的MSE绝对值最小,|MSE|≤0.15 eV;ωB97XD、CAM-B3LYP的偏差最大,|MSE|≥0.39 eV. 对于MAE,B3LYP、mPW3PBE、B3PW91最小,MAE≤0.19 eV;ωB97XD、CAM-B3LYP的MAE值最大,MAE≥0.53 eV. 对于RMS,B3LYP、mPW3PBE、B3PW91泛函最小,RMS≤0.24 eV;ωB97XD、CAM-B3LYP的最大,为0.79 eV. 对于SD,B3LYP、mPW3PBE和B3PW91的最小,SD≤0.17 eV;ωB97XD、CAM-B3LYP的最大,SD≥0.33 eV. 与表3中激发能统计数据分析比较,发现B3LYP、mPW3PBE、B3PW91 3种泛函计算垂直发射能的MAE、RMS、SD预测值均略小于激发能的对应值. 由表4中的数据可知,对于垂直发射能的计算,B3LYP、mPW3PBE、B3PW91 3种泛函能够给出较为精确的结果,这和垂直激发能的计算是一致的.
综上所述B3LYP、mPW3PBE、B3PW91 3种泛函在计算预测萤光类似物光谱性质方面有很好的表现,统计分析中的表现均名列前茅. 所以B3LYP、mPW3PBE、B3PW91在讨论萤光素类似物光谱性质方面是合适的选择.
2.3 基态和激发态几何结构的讨论
8种化合物基态到激发态构型列于图3. 从图3中可以看出基态分子二面角(苯并芳环与噻唑环之间的二面角)均为180°,激发态分子二面角介于178.3°~180°之间,说明8种物质基态、激发态苯并芳环和噻唑环之间均为共面,可以很好地形成共轭体系. 基态分子C1-C2键长为0.145 2~0.147 7 nm,略小于常规的C-C单键(0.153 nm),说明两环之间电子存在一定的共轭. 激发态时C1-C2键长缩短,这意味着在电子激发态时,噻唑环与左侧苯并芳环间电子离域性加强,C1-C2键增强. 此外,从基态到第一激发态,噻唑环C2-N、C2-S键变长. 激发态时所有物质噻唑环部分C3-N键变短,且4种未被氧化型萤光素类似物C3-N键长变化不超过0.001 nm,而4种氧化型萤光素类似物C3-N键长变化大于0.003 nm. 这可能是由于萤光类似物被氧化之后羧基变成羰基,从而C=O基团与噻唑环的C=N键共轭,使C3-N键长受电子激发过程影响更加明显. 基态到激发态所有8种萤光素类似物的苯并芳环环上取代基到环的键长缩短,在NAL、QAL、CAL、AL中C-NH2键长平均缩短0.002 7 nm,在OXYLH2、DHOXYLH2、DMOXYLH2、MDMOXYLH2中C-OH键长平均缩短0.002 4 nm.


图3 在B3LYP/6-31+G(d)计算水平下8种荧光素类似物基态和第一激发态(括号中)优化后的结构数据(键长:nm,角度:°) Fig.3 Optimized geometrical parameters of ground state and the first singlet excited state (in parentheses) of eight analogs of luciferin at B3LYP/6-31+G(d) level(bond: nm , angle: °)
2.4 NBO电荷分析
图4列出了8种萤光素类似物最高占据轨道(HOMO)和最低空轨道(LUMO)的分子轨道轮廓图. TD-DFT计算表明,所有这8种物质的S0态到S1态的跃迁均对应着一个电子从HOMO轨道向LUMO轨道的跃迁. 从图4可见,8种物质S0到S1均为π-π*跃迁. 由图4可见,8个结构的HOMO和LUMO轨道电子云重叠程度较大,意味着S0和S1态间的电子跃迁几率大,荧光发光效率也会较高. 此外,比较HOMO和LUMO轨道可以看出,随着8种萤光素类似物从S0态到S1态的跃迁,伴随着部分电荷从左侧苯并芳环向右侧噻唑环的转移. 如图4所示,HOMO和LUMO在苯并芳环与取代基(-NH2或-OH)键合处表现出明显的反键特征,随着分子从基态跃迁至激发态,电子密度从左向右转移,相当于苯并芳环与取代基(-NH2或-OH)间的反键轨道上的电子数减少,C-NH2或C-OH键强度增加,使得激发态分子中取代基到苯并环的共价键长缩短. 此外,对所有8种结构,LUMO在C2-S和C2-N(原子序号见图3)处表现出明显的反键特征,而对后4种结构,LUMO在C3-N处表现出明显的成键特征. 因此,随着HOMO→LUMO的电子跃迁,噻唑环上的C2-S,C2-N共价键的反键轨道上电子占据数增加,从而导致C2-N、C2-S键增长;而对后4种结构,C3-N键的成键轨道电子占据数增加,C3-N键缩短.

图4 8种萤光素类似物前线分子轨道示意图 Fig.4 Frontier orbitals for eight analogs of luciferin
为了探讨激发态时电荷的转移程度,对8种物质分别进行NBO电荷分析. 8种物质的NBO电荷列于表5中. 从表5可以看出,从S0态到S1态跃迁,8种物质发生一定程度的分子内电荷转移,电荷从苯并噻唑和取代基向噻唑环转移. 各物质电荷转移程度如下:AL(0.28 e),CAL(-0.03 e),NAL(0.25 e),QAL(0.19 e),DHOXYLH2(0.44 e),DMOXYLH2(0.34 e),MDMOXYLH2(0.35 e),OXYLH2(0.34 e). 可以观察到以上8种物质的ICT过程可分为两组:(1)未被氧化的萤光素类似物,其电荷转移程度较小,为-0.03 ~0.28 e;(2)氧化的萤光类似物,其电荷转移程度增大,为0.34~0.44 e. 可见氧化后的萤光素类似物的ICT整体大于未被氧化的萤光素类似物的.

表5 8种萤光素类似物噻唑环部分的基态、第一激发态的NBO电荷
荧光化合物发射光谱在溶液中的红移现象已得到广泛研究,红移大小与溶剂的极性以及化合物S1态发生的电荷转移的程度直接相关. 电荷转移越强烈,基态到激发态偶极矩变化越明显,则溶剂效应对发射光谱的影响越明显. 已有文献表明CAL发射光谱与溶剂的极性无关,AL、NAL、QAL的发射光谱随着溶剂极性的增加其光谱均发生不同程度的红移[29]. 由表5可见,CAL基态到激发态时发生的电荷转移程度很小(-0.03 e),所以其发射光谱受溶剂的影响很小;其他3种结构在S1态时均发生明显的分子内电荷转移(0.19~0.28 e),导致它们的发射光谱受溶剂的影响较大. 由于氧化萤光素类似物的S1态CT程度远大于氨基取代萤光素类似物,所以本文同样可以预测DHOXYLH2、DMOXYLH2、MDMOXYLH2、OXYLH2的发射光谱受溶剂的影响,随着溶剂极性增大发射光谱将发生红移. DREW等[41]曾对TD-DFT处理的分子内电荷转移时不同泛函的影响做过讨论和分析,他们指出随着分子内电荷转移程度增大,为了得到准确的激发能就需要在泛函中掺杂更多的HF交换能[24]. 本文的算例表明,8种物质均有不同程度的电荷转移,含有一定HF交换能比例的B3LYP、mPW3PBE、B3PW91泛函比纯泛函计算预测其光谱性能理想,验证了DREW等的结论. 因此只要对萤光物质的组成、结构(包括激发态CT的程度)进行适当的分类,选择不同的泛函,可以有效的提高TD-DFT计算预测萤光类似物光谱性质的准确性.
3结论
采用10种不同的DFT泛函在DMSO溶液中对8种萤光素类似物(NAL、QAL、AL、CAL、OXYLH2、DHOXYLH2、DMOXYLH2、MDMOXYLH2)吸收光谱和发射光谱进行了预测,同时分析了各个物质基态到激发态时结构和电荷的变化. 结果表明DFT泛函对于萤光素类似物光谱性能的预测可以给出比较合理的结果,B3LYP、mPW3PBE、B3PW91三种泛函计算预测的发射能RMS≤0.24 eV,与实验值吻合较好. 另外两种H-GGA泛函PBE0、B3P86的统计误差相较于其他泛函也较小,纯泛函和长程校正的泛函的误差均很大. 本文最终选取的三种H-GGA的方法中mPW3PBE、B3PW91在前期文献讨论其他荧光素类似物结构中也被推荐,说明这两种方法在研究该体系具有较为广泛的适用性. 希望本文能够对以后研究讨论萤光素类似物方面尤其是理论计算方面提供一些帮助.
参考文献:
[1] 孙颖, 任爱民, 李作盛, 等. 海萤荧光素类似物发光反应机理的理论研究[J]. 高等学校化学学报, 2011, 32(11): 2586-2592.
[2] 李作盛, 邹陆一, 任爱民, 等. 萤火虫氧化荧光素类似物电子光谱的理论研究[J]. 高等学校化学学报, 2012, 33(12): 2757-2764.
[3] LI Z S, MIN C G, REN A M, et al. TD-DFT investigation of fluorescence properties of luciferin and oxyluciferin analogs bearing an amino group [J]. J Photochem Photobiol A: Chem, 2012, 243: 7-16.
[4] VIVIANI V A. The origin, diversity, and structure function relationships of insect luciferases [J]. Cell Mol Life Sci, 2002, 59(11): 1833-1850.
[5] NAVIZET I, LIU Y J, FERRÉ N, et al. Color-tuning mechanism of firefly investigated by multi-configurational perturbation method [J]. J Am Chem Soc, 2010, 132(2): 706-712.
[6] CAI D J, MARQUES M A L, NOGUEIRA F. Accurate color tuning of firefly chromophore by modulation of local polarization electrostatic fields [J]. J Phys Chem B, 2011, 115(2): 329-332.
[7] ANDO K, NIWA K, YAMADA N, et al. Firefly bioluminescence quantum yield and colour change by pH-sensitive green emission [J]. Nat Photonics, 2008(2): 44-47.
[8] ORLOVA G, GODDARD J D, BROVKO L Y. Theoretical study of the amazing firefly bioluminescence:the formation and structures of the light emitters [J]. J Am Chem Soc, 2003, 125(23): 6962-6971.
[9] BRANCHINI B R, MURTIASHAW M H, MAGYAR R A, et al. Yellow-green and red firefly bioluminescence from 5,5-dimethyloxyluciferin [J]. J Am Chem Soc, 2002, 124(10): 2112-2113.
[10] HIRANO T, HASUMI Y, OHTSUKA K, et al. Spectroscopic studies of the light-color modulation mechanism of firefly (beetle) bioluminescence [J]. J Am Chem Soc, 2009, 131(6): 2385-2396.
[11] HOSSEINKHANI S. Molecular enigma of multicolor bioluminescence of firefly luciferase [J]. Cell Mol Life Sci, 2011, 68(7): 1167-1182.
[12] NAUMOV P, OZAWA Y, OHKUBO K, et al. Structure and spectroscopy of oxyluciferin, the light emitter of the firefly bioluminescence [J]. J Am Chem Soc, 2009, 131(32): 11590-11605.
[13] NAUMOV P, KOCHUNNOONNY M. Spectral-structural effects of the keto-enol-enolate and phenol-phenolate equilibria of oxyluciferin [J]. J Am Chem Soc, 2010, 132(33): 11566-11579.
[14] CHEN S F, YUE L, LIU Y, et al. Multireference theoretical studies on the solvent effect of firefly multicolor bioluminescence [J]. J Quantum Chem, 2011, 111(13): 3371-3377.
[15] LIU F Y, LIU Y J, VICO L D, et al. Theoretical study of the chemiluminescent decomposition of dioxetanone [J]. J Am Chem Soc, 2009, 131(17): 6181-6188.
[16] CHEN S F, LIU Y J, NAVIZET I, et al. Systematic Theoretical investigation on the light emitter of firefly [J]. J Chem Theory Comput, 2011, 7(3): 798-803.
[17] MARQUES S M, ESTEVES DA SILVA J C G. Firefly bioluminescence: a mechanistic approach of luciferase catalyzed reactions [J]. IUBMB Life, 2009, 61(1): 6-17.
[19] PINTO DA SILVA L, ESTEVES DA SILVA J C G. Computational studies of the luciferase light-emitting product: oxyluciferin [J]. J Chem Theory Comput, 2011, 7(4): 809-817.
[20] VIVIANI V R, ARNOLDI F G C, NETO A J S, et al. The structural origin and biological function of pH-sensitivity in firefly luciferases [J]. Photochem Photobiol Sci, 2008, 7(2): 159-169.
[21] 韩洁, 李剑利, 贺怀贞, 等. 新型荧光素类细胞钙离子荧光探针Fluo-Cl的设计、合成及表征[J]. 高等学校化学学报, 2008, 29(10): 2003-2006.
[22] 李丙瑞, 何凤英, 王流芳. 荧光素衍生物抗癌活性的模式识别[J]. 高等学校化学学报, 1993, 14(7): 954-956.
[23] 李晓花, 马会民, 董素英, 等. 一种新的荧光素类荧光探针的设计合成及其对血清中组氨酸的选择性标记[J]. 高等学校化学学报, 2003, 24(11): 1984-1986.
[24] 王溢磊, 吴国是. 香豆素衍生物的荧光发射能计算及XC泛函的合理选择[J]. 物理化学学报, 2007, 23(12): 1831-1838.
[25] 宋争林, 张复实, 陈锡侨, 等. 酞菁基态和激发态的计算[J]. 物理化学学报, 2003, 19(2): 130-133.
[26] SUNDHOLM D. Density functional theory calculations of the visible spectrum of chlorophyll [J]. Chem Phys Lett, 1999, 302(5/6): 480-484.
[27] DREUW A, WEISMAN J L, HEAD-GORDEN M. Long-range charge-transfer excited states in time-dependent density functional theory require non-local exchange [J]. J Chem Phys, 2003, 119(6): 2943-2946.
[28] SALZNER U. Theoretical investigation of excited states of large [J]. J Chem Theory Comput, 2007, 3(1): 219-231.
[29] VIEIRA J, DA SILVA L P, ESTEVES DA SILVA J C G. Advances in the knowledge of light emission by firefly luciferin and oxyluciferin [J]. J Photochem Photobiol B, 2012, 117: 33-39.
[30] BECKE A D. Density-functional exchange-energy approximation with correct asymptotic behavior [J]. Phys Rev A, 1988, 38(6): 3098-3100.
[31] LEE C, YANG W T, PARR R G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density [J]. Phys Rev B, 1988, 37(2): 785-789.
[32] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple [J]. Phys Rev Lett, 1996, 77(18): 3865-3868.
[33] ADAMO C, BARONE V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models [J]. J Chem Phys, 1998, 108(2): 664-675.
[34] PERDEW J P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas [J]. Physl Rev B, 1986, 33(12): 8822-8824.
[35] BECKE A D. Density-functional thermochemistry. iv. a new dynamical correlation functional and implications for exact-exchange mixing [J]. J Chem Phys, 1996, 104(3): 1040-1046.
[36] YANAI T, TEW D P, HANDY N C. A new hybrid exchange-correlation functional using the coulomb-attenuating method (cam-b3lyp) [J]. Chem Phys Lett, 2004, 393(1/3): 51-57.
[37] CHAI J D, HEAD-GORDON M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections [J]. Phys Chem Chem Phys, 2008, 10(44): 6615-6620.
[38] JACQUEMIN D, PLANCHAT A, ADAMO C, et al. TD-DFT assessment of functionals for optical 0-0 transitions in solvated dyes [J]. J Chem Theory Comput, 2012, 8(7): 2359-2372.
[39] GLENDENING E D, REED A E, CARPENTER J E, et al. NBO [CP]. Version 3.1, 2010.
[40] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09 [CP]. Revision C. 01, Wallingford CT: Gaussian Inc., 2010.
[41] DREUW A, DUNIETZ B D, HEAD-DORDON M. Characterization of the relevant excited states in the photodissociation of co-ligated hemoglobin and myoglobin [J]. J Am Chem Soc, 2002, 124(41): 12070-12071.
[责任编辑:吴文鹏]
猜你喜欢
昆钢科技(2022年2期)2022-07-08
昆钢科技(2021年4期)2021-11-06
分析化学(2018年12期)2018-01-22
中国资源综合利用(2017年1期)2018-01-22
环境保护与循环经济(2017年3期)2017-09-26
山东工业技术(2016年15期)2016-12-01
现代食品(2016年14期)2016-04-28
当代化工研究(2016年9期)2016-03-20
中国粮油学报(2016年5期)2016-01-23
油气田环境保护(2015年4期)2015-12-28
