
胰腺神经内分泌肿瘤肝转移合并异位ACTH 综合征1 例报道
2015-12-31 09:12程捷瑶阮戈冲
胃肠病学和肝病学杂志 2015年12期
张 巍,程捷瑶,阮戈冲,孙 菲,杨 红
1.中国医学科学院 北京协和医院 1.内科;2.消化内科,北京100730
胰腺神经内分泌肿瘤(pancreatic neuroendocrine tumor,pNET)是一种少见的胰腺肿瘤,年发病率约1/10万,根据是否伴随相应的内分泌症状可分为功能性和无功能性两类,其中功能性pNET 以胰岛素瘤、胃泌素瘤两种类型多见,也有胰高血糖素瘤、血管活性肠肽瘤、生长抑素瘤等少见类型。本文就北京协和医院(下称“我院”)消化内科收治pNET 肝转移合并异位ACTH 综合征1 例诊治过程探讨如下。
病例 患者,女,49 岁,2008 年4 月无明显诱因出现腹泻,水样便,4 ~10 次/d。当地医院就诊,以“急性胃肠炎”行抗感染治疗,效果欠佳,后行胃镜检查提示“糜烂性胃炎、十二指肠溃疡”,予兰索拉唑、匹维溴铵、黄连素等药物后症状缓解,停药后病情反复。2009 年11 月收住我院,查:血清胃泌素138 pg/ml,血尿便常规、便寄生虫检查、肝肾功、甲功、24 h 尿游离皮质醇、NSE、甲状旁腺素、肿瘤标记物均无异常。胃镜示反流性食管炎、十二指肠炎;超声内镜示胰腺体尾部占位;生长抑素受体显像示左侧肾上腺生长抑素高表达病变;腹盆增强CT +胰腺灌注提示:胰尾高强化病变。结合患者临床症状、影像学和内镜表现,诊断考虑为pNET。2009 年12 月于全麻下行胰体尾及脾脏切除术,术中见胰腺体尾部近脾门处约4.0 cm ×3.0 cm 大小实性肿物,质硬,边界尚清晰。术后病理示:生物学行为不确定胰岛细胞瘤(胃泌素瘤),核分裂<1/10 HPF,血管内有瘤栓,淋巴结未见转移。免疫组化:CgA(+),Syn(+),Gastrin(+),Insnlin(-),Glucagon(-),Somatostatin(-),CD34(血管+),Ki-67 约5%。术后恢复顺利,出院后规律口服耐信、得每通,无特殊不适。2011 年7 月复查血清胃泌素33.6 pg/ml;腹部增强CT 示pNET 行胰体尾脾脏切除术后改变,肝右叶新发多发异常强化灶,生长抑素受体显像未见异常(见图1)。2012 年9 月复查胃泌素123.7 pg/ml;腹部增强CT 示肝脏多发占位增多增大,胰头较前增大(见图2)。患者未进一步治疗。2013 年3 月起出现多饮、多食、多尿,伴口干、大汗、心悸,查空腹血糖>11.1 mmol/L,患者未诊治。2013 年8 月查血清胃泌素62.8 pg/ml,NSE 19.2 ng/ml;肝功ALT 93 U/L,AST 61 U/L,ALP 160 U/L,GGT 346 U/L;腹部增强CT 示肝脏多发占位较前进一步增大;生长抑素受体显像示新增肝内多发生长抑素受体高表达病变,部分较大病灶中心液化坏死,腹膜后数枚小淋巴结,生长抑素受体有轻度表达(见图3)。建议予生长抑素+肝内血管栓塞治疗,患者拒绝。2013 年10 月患者出现满月脸、双颊冻疮样皮疹,面部及双下肢胫前轻度可凹性水肿,血压最高160/80 mm-Hg,空腹血糖最高26 mmol/L,当地医院予速尿、螺内酯、厄贝沙坦氢氯噻嗪、诺和锐治疗,血压血糖控制可,水肿无明显好转,出院后仍间断水肿,未及时就诊。2014 年3 月面部水肿加重,双下肢水肿蔓延至腹部,利尿效果差。2014 年5 月第2 次入我院查血清CgA 600 U/L;大剂量地塞米松抑制试验示不能抑制;腹部B 超、腹盆增强CT、肾上腺CT 均提示肝脏多发占位,转移瘤可能,肝动脉分支参与病灶供血,门脉、肝静脉、下腔静脉管腔局部受压,左肾上腺增粗;鞍区MRI 未见异常;生长抑素受体显像示肝内神经内分泌转移瘤较前增大,部分内部伴液化坏死。考虑ACTH 依赖性库欣综合征,异位ACTH 来源可能性大。遂取2009 年胰腺手术病理行ACTH 及CRH 染色,ACTH局灶(+)(见图4 ~5)。考虑肝占位为pNET 肝脏转移,建议患者行肾上腺切除或肝动脉栓塞治疗或长效奥曲肽治疗。患者因经济原因拒绝使用长效奥曲肽,故分别于2014 年6 月、7月、9 月在局麻下行3 个疗程的肝动脉栓塞介入治疗,术程均顺利,术后予善宁0.1 mg q8 h 皮下注射×2 d 及保肝对症治疗。2014 年9 月复查:血皮质醇:37.79 μg/dl(术前)→5.07 μg/dl(术后);24 h 尿皮质醇:858 μg/24 h(术前)→3.04 μg/24 h(术后);血ACTH:152.0 pg/ml(术前)→64.9 pg/ml(术后);胸腹盆增强CT 示:胸部未见明显异常;肝脏多发占位较前缩小。现患者无发热、水样便、脓血便、便中带油花,无反酸、烧心、恶心、呕吐,库欣综合征表现较前明显减轻。

图1 腹部增强CT 示胰尾高强化病变;图2 腹部增强CT 示肝脏多发占位Fig 1 Abdominal contrast-enhanced CT scanning showed highly strengthened lesions in tail of pancreas;Fig 2 Abdominal contrast-enhanced CT scanning showed multipleoccupying lesions in the liver
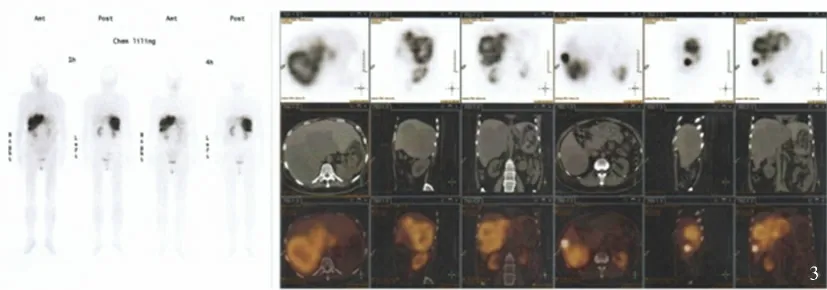
图3 生长抑素受体显像示肝内多发生长抑素受体高表达病变Fig 3 Somatostatin receptor scintigraphy showed multiple SSTR high-expression lesions in the liver

图4 胰体尾切除术后病理(HE 150 ×);图5 胰腺手术病理行ACTH 及CRH 免疫组化染色,ACTH 局灶(+)(HE 150×)Fig 4 Postoperative histopathological results after partial pancreatectomy (HE 150 ×);Fig 5 Immunohistochemical staining of ACTH and CRH after partial pancreatectomy,showed ACTH-positive lesions (HE 150 ×)
讨论 pNET 是一种少见肿瘤,仅占全部胰腺肿瘤的1%~2%,在全部消化系统神经内分泌肿瘤中仅占约12.1%[1]。pNET 又分为功能性和非功能性,功能性pNET 常有特异性临床表现。对于功能性pNET 的血清学检测应包括相应激素及其代谢产物,如本例中血皮质醇及血ACTH 的水平变化很好地反映了肿瘤转移、进展及治疗情况。此外,非特异性的肿瘤标记物如嗜铬粒蛋白A(chromogranin A,CgA)等也常用于pNET 的诊断。血清CgA 可表达于约90%的pNET 病例中,且其水平与肿瘤负荷、对治疗的反应密切相关,因此,目前被认为是最敏感的pNET 血清肿瘤标记物[2]。本例患者第2 次入院时血清CgA明显升高,与pNET 复发转移的病情一致。
本例患者第2 次入院时ACTH 异常增高,通过检查垂体、肾上腺等提示为异位ACTH 分泌。异位ACTH 综合征(ectopic ACTH syndrome,EAS)指由垂体以外的组织分泌大量ACTH 导致临床上出现库欣综合征的表现。根据文献资料,合并有EAS表现的pNET 分别占全部pNET 病例和全部EAS 病例的1.2%和3.6%[3]。其发生率虽低,但通常恶性程度较高,病情进展迅猛,多数早期既转移至肝脏[4]。本例中,胰腺原发灶ACTH染色既已为阳性,但患者仅表现为胃泌素瘤相关临床症状,而无EAS 表现,直至术后3 年明确pNET 肝脏转移后,方有明显的EAS 表现即库欣综合征出现。推测其可能的原因,其一在于EAS 出现相对缓慢,常被生长迅速的pNET 原发灶表现掩盖;其二在于pNET 在向肝脏转移的过程中极可能出现了去分化改变,肿瘤分化变低,恶性程度升高,外在表现为分泌激素类型的变化。查阅国内外文献也可见类似个案报道,即患者原发肿瘤的ACTH 及皮质醇染色均为阴性,但肝脏转移灶显示ACTH 强阳性,患者出现EAS 临床表现[5]。pNET 的这个特性提示我们应当充分重视其肿瘤细胞的多潜能性,以动态的眼光看待pNET 及其转移灶的组织病理学、内分泌学特征。必要时重复组织活检及免疫组化检查对于调整诊断和治疗方案、评估疾病预后均有重要意义。
pNET 的治疗可通过手术切除、化疗等手段以减轻肿瘤负荷、改善临床症状。值得注意的是,在功能性pNET 的治疗中,全程严格控制由肿瘤释放的过量激素所引起的内分泌症状尤为重要,因为既往研究已指出这些内分泌症状常成为致死的首要原因。本例中,早期切除胰体尾部肿瘤、明确肿瘤肝转移后积极进行肝动脉栓塞治疗即有减少肿瘤内分泌的考量。此外,越来越多证据表明生长抑素类似物可以通过pNET 肿瘤细胞表面的生长抑素受体,尤其是SSTR-2、SSTR-3 和SSTR-5 3 种亚型,不仅能控制肿瘤细胞分泌过量激素,还能直接抑制肿瘤细胞生长[6]。本例患者因个人原因仅在介入围手术期应用了短效奥曲肽,治疗上仍取得部分疗效。尽管无法分辨每项治疗措施的具体效果,但综合运用手术、动脉栓塞、生长抑素措施在本例中有效控制了内分泌症状及肿瘤进展。根据文献资料,有远处脏器转移的G1期、G2期pNET 患者中位生存时间为33 个月,5 年生存率约35%;而有远处脏器转移的G3期pNET 患者中位生存时间仅为5 个月,5 年生存率不足5%[7]。与之相比,本例患者原发病灶Ki-67 指数5%,考虑为G2期,自发现pNET肝脏多发转移至今已3 年余,且目前症状控制良好,总体疗效基本满意。
综上,本例中的pNET 肿瘤细胞充分显示了其多潜能性,在生长及转移过程中可能出现了去分化改变,导致分泌激素的类型产生明显变化。合并有EAS 表现的pNET 临床少见,但通常恶性程度较高,病情进展迅猛,综合运用手术、动脉栓塞、生长抑素等多种治疗手段控制内分泌症状、减轻肿瘤负荷,可有效延长患者生存期。
[1] Cives M,Strosberg J. An update on gastroenteropancreatic neuroendocrine tumors[J]. Oncology (Williston Park),2014,28 (9):749-756.
[2] Strosberg JR,Nasir A,Hodul P,et al. Biology and treatment of metastatic gastrointestinal neuroendocrine tumors[J]. Gastrointest Cancer Res,2008,2(3):113-125.
[3] Ito T,Tanaka M,Sasano H,et al. Preliminary results of a Japanese nationwide survey of neuroendocrine gastrointestinal tumors[J]. J Gastroenterol,2007,42(6):497-500.
[4] Jensen RT,Cadiot G,Brandi ML,et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms:functional pancreatic endocrine tumor syndromes[J]. Neuroendocrinology,2012,95(2):98-119.
[5] Miehle K,Tannapfel A,Lamesch P,et al. Pancreatic neuroendocrine tumor with ectopic adrenocorticotropin production upon second recurrence[J]. J Clin Endocrinol Metab,2004,89(8):3731-3736.
[6] Miljkovic' MD,Girotra M,Abraham RR,et al. Novel medical therapies of recurrent and metastatic gastroenteropancreatic neuroendocrine tumors[J]. Dig Dis Sci,2012,57(1):9-18.
[7] Yao JC,Hassan M,Phan A,et al. One hundred years after“carcinoid”:epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States[J]. J Clin Oncol,2008,26(18):3063-3072.
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
心电与循环(2021年4期)2021-11-29
智慧健康(2021年33期)2021-03-16
康颐(2020年8期)2020-11-03
天津医科大学学报(2019年3期)2019-08-13
中学数学杂志(2019年9期)2019-05-29
中国医药指南(2017年3期)2017-11-13
现代检验医学杂志(2016年3期)2016-11-15
中国眼镜科技杂志(2016年17期)2016-10-24
中国卫生标准管理(2015年8期)2016-01-15
