
绿色杀菌剂肟菌酯的合成
2015-12-28 07:35金朝俊
徐州工程学院学报(自然科学版) 2015年3期
关键词:合成
袁 宇,金朝俊,刘 洋
(扬州大学 化学化工学院,江苏 扬州 225002)
绿色杀菌剂肟菌酯的合成
袁宇,金朝俊,刘洋
(扬州大学 化学化工学院,江苏 扬州225002)
摘要:肟菌酯是一种广谱、高效、低毒而且对环境友好的嗜球果伞素类杀菌剂,其市场前景广阔.肟菌酯因含有E型的β-甲氧基丙烯酸甲酯的结构而具有杀菌效果,研究发现其作用机理是通过阻断真菌体内的呼吸通道来完成的.运用逆推法探索分别以邻甲基苯乙酮和邻甲基苯甲酸为原料探索了两条路线来合成重要中间体2-甲基-α-羰基苯乙酸甲酯.再使其与甲氧基胺盐酸盐肟化后,用NBS溴化生成(E)-2-溴代甲基-α-甲氧亚胺基苯乙酸甲酯,最后与(E)-间三氟甲基苯乙酮肟缩合生成肟菌脂,总收率为25.7%.
关键词:绿色杀菌剂;逆推法;肟菌酯;合成
肟菌酯 (Trifloxystrobin)是嗜球果伞素类杀菌剂类的一种[1],随着此类天然β-甲氧基丙烯酸酯衍生物的发现,人们研发出了新一类广谱、高效、低毒的杀菌剂.肟菌酯的开发为世界农药市场注入了新鲜的活力,该杀菌剂性能卓越、防效持久而且杀菌方式独特[2],所以其市场潜力巨大.另外,Bostanian等[3]通过大量动物实验和研究,发现肟菌酯毒性极小,如若被吸收进体内,大部分都会被排出体外,只会有极小的残留.对肟菌酯进行药理实验,发现它对胎仔无催畸形性,无突变异性,所以该杀菌剂安全性高.而且肟菌酯溶解快、易水解,也能在植物体内、土壤以及水中迅速降解,对动植物毒性极低,对环境友好,堪称绿色杀菌剂.
1肟菌酯传统合成工艺的概述
1993年Brand等[4]申请了一篇专利,专利中以邻甲基苯乙酸甲酯为原料,使用液溴溴化得到邻溴甲基苯乙酸甲酯,再让它与(E)-间三氟甲基苯乙酮肟反应得到(E)-2-(α-(((α-甲基-3-三氟甲基苯基)亚胺)氧)-O-甲苯基)-乙酸甲酯,接着再将其氧化,之后与甲氧基胺盐酸盐反应,最后得到目标产物肟菌酯.对于第一步反应,有文献选择溴化,也有文献选择氯化,因为氯代在工业上比较便宜,但是氯的离去性显然没有溴好,不易发生取代反应.Cliff等就使用NBS在四氯化碳中进行溴化反应,产率也比较理想.

1995年Ziegler等[5]以邻溴甲基苯硼酸为起始原料,将它跟(E)-间三氟甲基苯乙酮肟进行反应,得到相应的取代产物后再和2-氯-2-甲氧基亚胺-乙酸甲酯反应,即可得到肟菌酯.该方法最后一步使用到了昂贵的催化剂Pd(PPh3)4,成本较高,不适合工业化.

1996年Pfiffner等[6]使用邻溴甲基苯乙酸甲酯与苯甲羟酸作为起始原料,在碱性条件下脱去一分子HBr,产物再进一步在盐酸作用下得到邻氨基氧-甲基-苯乙酸甲酯的盐酸盐,之后,将其与间三氟苯乙酮脱去HCl和H2O,产品再与亚硝酸叔丁酯反应得到一个酮肟化合物,接着用硫酸二甲酯甲基化就可以得到最终产品.
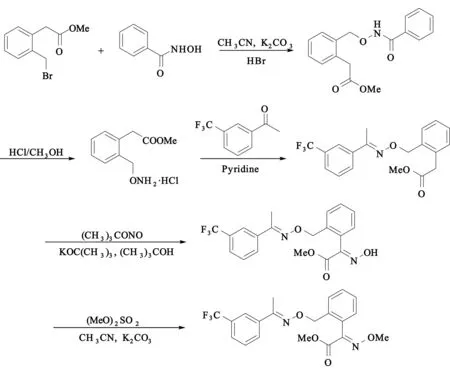
1998年Assercq等[7]以N, N-二甲基苄胺和草酸二甲酯作为起始原料,经过3步反应得到(E)-2-氯甲基-α-甲氧亚胺基苯乙酸甲酯,这3步总收率可达55%,之后再与(E)-间三氟甲基苯乙酮肟反应得到最终产物.该方法第一步需要使用正丁基锂,价格昂贵且危险性较大,不适合工业化.

2005年李焰等[8]又以邻溴甲苯和乙二酰氯单甲酯为原料,通过有机铜锂试剂进行偶联反应得到相应的酮酯,接着再与甲氧基胺盐缩合就可以得到一个十分重要的中间体肟酯,之后使用NBS进行溴化,产物与(E)-间三氟甲基苯乙酮肟反应制得肟菌酯.

Strobilurin类杀菌剂的优异性能已经使得它的研发成为了全球农药研发的热点[9].但是传统工艺合成路线繁琐、反应条件苛刻难以控制、产生大量的工业废水、废气、废渣给环境带来了巨大的压力.因此,我们探索了一条简单易行、经济环保的合成工艺路线.
2实验部分
2.1 仪器与试剂
仪器:85-1型磁力搅拌器;ZF-1型三用紫外仪;SHZ-D(III)型循环水式多用真空泵;DHG-9003型电热恒温鼓风干燥箱;R-3型旋转蒸发仪;气质联用仪型号为Fennigan LCQ Deca XP MAX;1H NMR和13C NMR使用的仪器型号为Buker AVANCE 600,溶剂为CDCl3,TMS作为内标物.
试剂:邻甲基苯甲酸、间三氟甲基苯乙酮均购于阿拉丁,邻甲基苯乙酮、甲氧基胺盐酸盐均购于安耐吉,N-溴代丁二酰亚胺、过氧苯甲酰、二氯亚砜、甲醇、乙醇、四氯化碳、溴化钠、浓硫酸、DMF、盐酸羟胺、氢氧化钠、高锰酸钾、亚硫酸氢钠、碳酸氢钠均购于国药集团,氰化钠是从江苏省长青农化股份有限公司取得,柱层析使用300~400目硅胶,TLC使用GF254高效薄层层析硅胶板.
2.2 合成方法
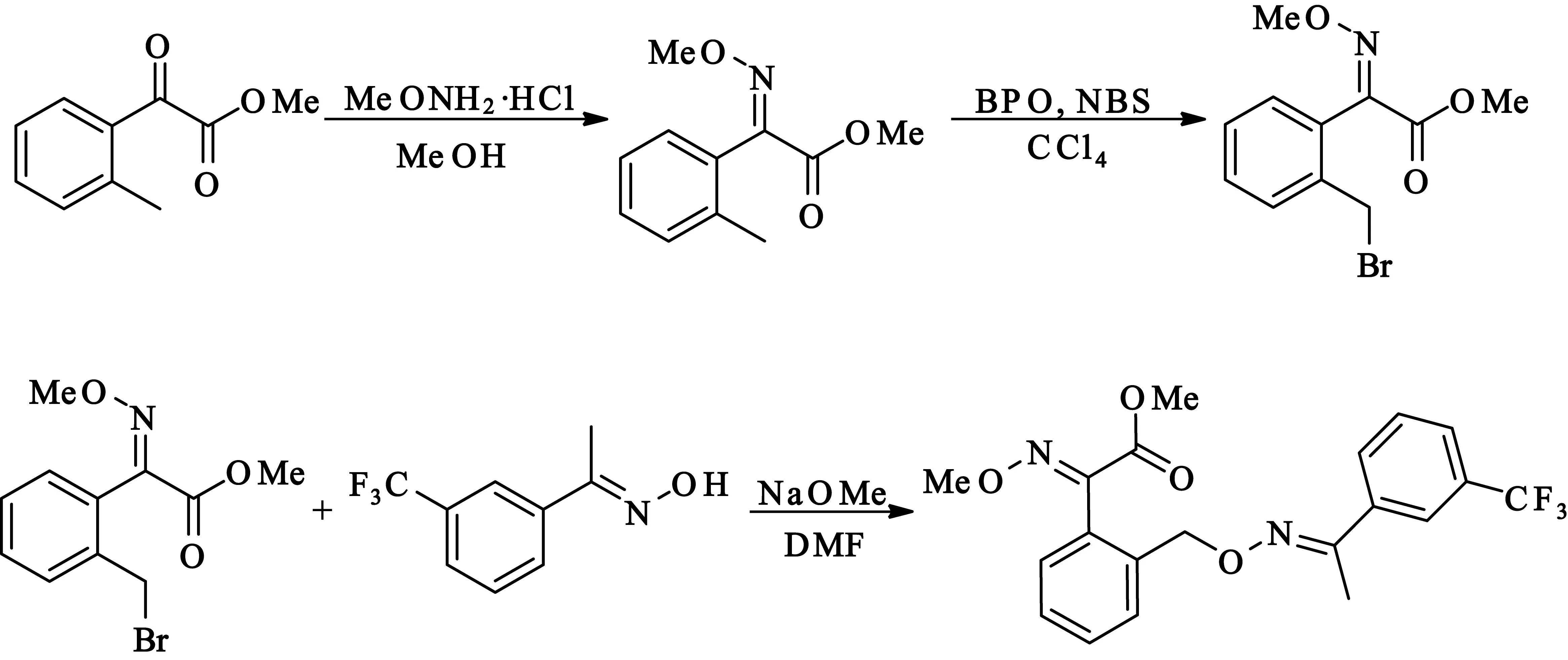
确定了从2-甲基-α-羰基苯乙酸甲酯合成肟菌脂的路线,将2-甲基-α-羰基苯乙酸甲酯与甲氧基胺盐酸盐在甲醇中回流得到(E)-2-甲基-α-甲氧亚胺基苯乙酸甲酯,然后,在四氯化碳中使用N-溴代丁二酰亚胺对2位甲基进行溴化生成(E)-2-溴代甲基-α-甲氧亚胺基苯乙酸甲酯,接下来在DMF和甲醇钠的条件下与(E)-间三氟甲基苯乙酮肟缩合生成肟菌脂.
2.2.12-甲基-α-羰基苯乙酸甲酯合成路线一

在500 mL圆底烧瓶中加入2 g (0.015 mol) 邻甲基苯乙酮、1 g 氢氧化钠、200 mL水,冰浴中搅拌下,将3.5 g (0.02 mol) 高锰酸钾分批加入烧瓶中,到最后一批高锰酸钾加入,反应体系中的颜色持续保持1 h 不变.反应结束,滴加亚硫酸氢钠溶液使混合液的紫红色褪去,接着,抽滤除去二氧化锰固体,水溶液用乙酸乙酯萃取除去未反应的原料.水相用浓盐酸酸化到pH = 1,有白色固体出现,抽滤,干燥,之后将所得固体加入到放有40 mL甲醇的100 mL圆底烧瓶中,搅拌下,缓缓滴加0.05 mL 浓硫酸,TLC跟踪反应.反应结束后用饱和的碳酸氢钠溶液调pH值至中性,再用乙酸乙酯萃取,合并有机层,无水硫酸钠干燥,蒸去乙酸乙酯后得到产品,2.2 g 黄色油状的2-甲基-α-羰基苯乙酸甲酯液体,产率83%.
2.2.22-甲基-α-羰基苯乙酸甲酯合成路线二
1)2-甲基苯甲腈的合成
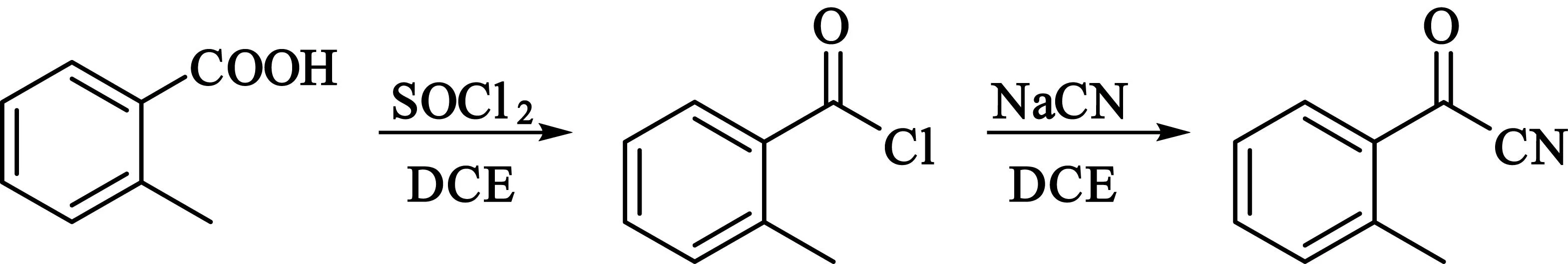
在500 mL的三口烧瓶中加入30 g(0.22 mol)邻甲基苯甲酸与200 mL 1,2-二氯乙烷,冰浴中缓慢滴加40 g(0.34 mol)二氯亚砜,滴毕后,加热回流14 h,蒸去剩余的二氯亚砜以及溶剂,即得到邻甲基苯甲酰氯32 g.将11.3 g(0.23 mol)NaCN溶于40 mL水中,倒入500 mL的三口烧瓶中,室温下加入0.7 g(0.002 mol)四丁基溴化铵以及200 mL 1, 2-二氯乙烷,剧烈搅拌,再缓慢滴加所制得邻甲基苯甲酰氯,室温反应16 h,反应结束后用1,2-二氯乙烷萃取,水洗,干燥,得到红棕色油状物,蒸馏纯化,得到淡黄色纯品24 g,产率为80%.
1H NMR(600 MHz,CDCl3):δ=2.60(s,3H),3.96(s,3H),7.30~7.33(m,2H),7.49(t,J=7.5 Hz,1H),7.68(d,J=7.8 Hz,1H)
13C NMR(150 MHz,CDCl3):δ=21.1,52.5,125.8,131.4,132.0,132.3,133.5,141.1,164.8,188.4
MS (EI):m/z=178[M+].
2)2-甲基-α-羰基苯乙酸甲酯的合成
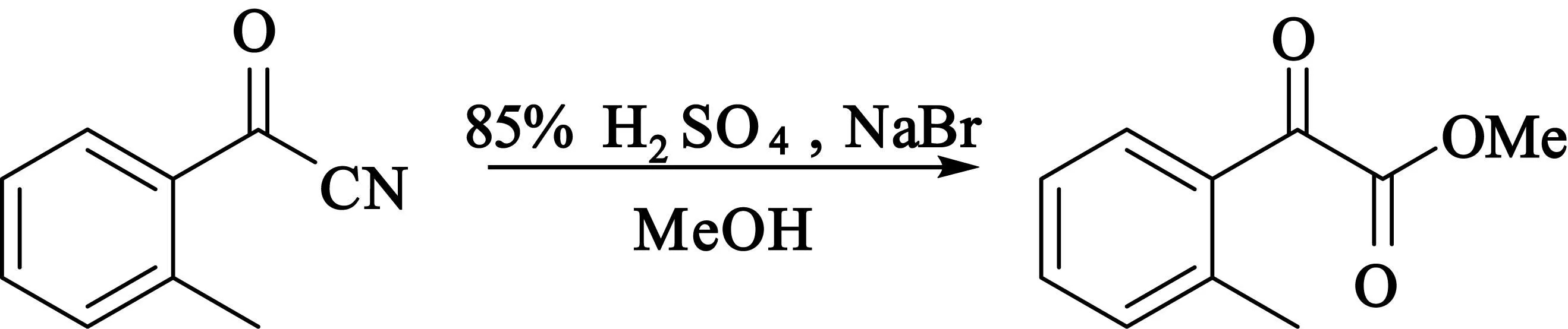
在50 mL的圆底烧瓶中加入85%硫酸(171 mmol 浓硫酸)和46.35 mg(0.45 mmol)溴化钠,室温搅拌下缓慢滴加1.305 g(9 mmol)邻甲基苯甲酰腈,滴加完毕,45 ℃搅拌1 h后加入9 mL甲醇,再搅拌3 h.反应结束后,加水、乙酸乙酯淬灭,萃取,干燥,蒸去乙酸乙酯后的残留液经硅胶柱层析纯化,得到1.169 g 2-甲基-α-羰基苯乙酸甲酯,产率73%.
1H NMR(600 MHz,CDCl3):δ=2.64(s,3H),7.34(d,J=7.6 Hz,1H),7.48(t,J=7.6 Hz,1H),7.63(t,J=7.5 Hz,1H),8.24(d,J=7.9 Hz,1H)
13C NMR(150 MHz,CDCl3):δ=18.9,116.0,126.4,129.9,132.1,134.4,137.5,139.5,199.4
MS (EI):m/z=145[M+].
2.2.3(E)-2-甲基-α-甲氧亚胺基苯乙酸甲酯的合成
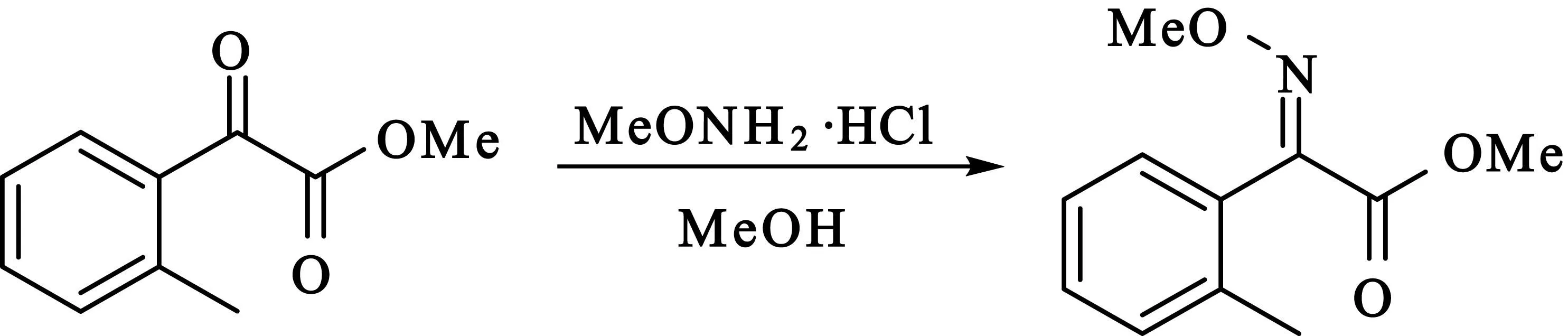
在50 mL三颈瓶中加入695 mg(3.9 mmol)2-甲基-α-羰基苯乙酸甲酯、391 mg(4.7 mmol)甲氧基胺盐酸盐和15 mL无水甲醇,TLC跟踪反应,搅拌回流14 h之后倒入水中,用乙酸乙酯萃取,无水硫酸钠干燥有机相,蒸去乙酸乙酯后的残留液经硅胶柱层析纯化,得到510 mg(E)-2-甲基-α-甲氧亚胺基苯乙酸甲酯晶体,产率62%.
1H NMR(600 MHz,CDCl3):δ=2.60(s,3H),3.96(s,3H),7.30~7.33(m,2H),7.49(t,J=7.5 Hz,1H),7.68(d,J=7.8 Hz,1H)
13C NMR(150 MHz,CDCl3):δ=21.1,52.5,125.8,131.4,132.0,132.3,133.5,141.1,164.8,188.4
MS(EI):m/z=178[M+].
2.2.4(E)-2-溴甲基-α-甲氧亚胺基苯乙酸甲酯的合成
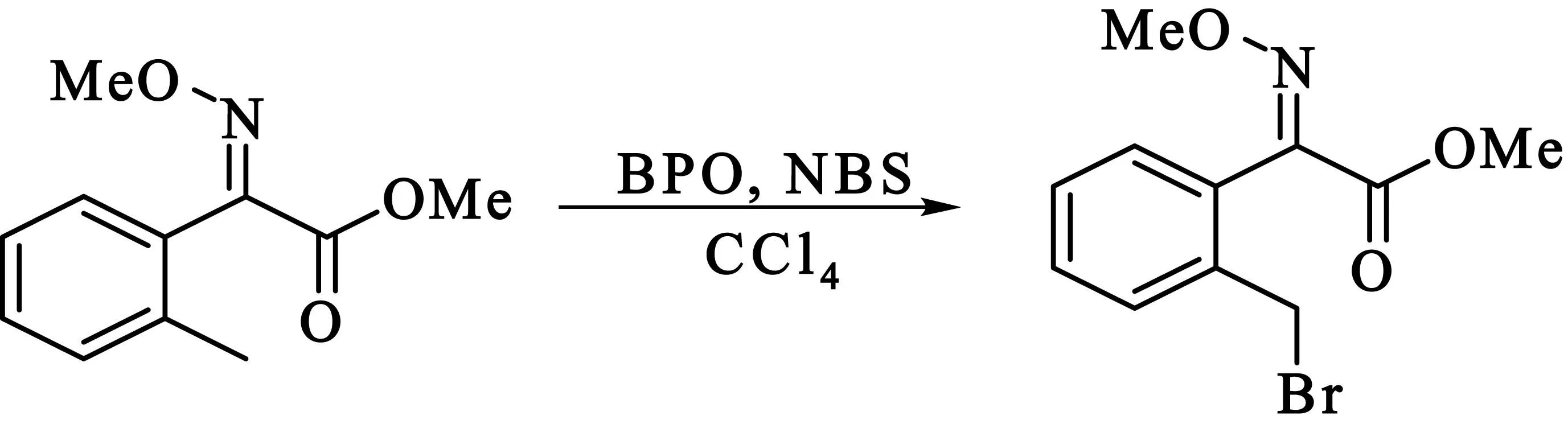
480 mg(2.32 mmol)(E)-2-甲基-α-甲氧亚胺基苯乙酸甲酯溶于10 mL四氯化碳置于50 mL圆底烧瓶中,加入578 mg(3.25 mmol)N-溴丁二酰亚胺和48 mg过氧苯甲酰.TLC跟踪反应进程,反应结束后乙酸乙酯萃取,有机相用无水硫酸钠干燥,蒸出溶剂,再经硅胶柱层析纯化,得538.5 mg无色(E)-2-溴甲基-α-甲氧亚胺基苯乙酸甲酯液体,产率为81%.
1H NMR(600 MHz,CDCl3):δ=2.23(s,3H),3.90(s,3H),4.09(s,3H),7.14(d,J=7.5 Hz,1H),7.29~7.29(m,2H),7.35(t,J=7.5 Hz,1H)
13C NMR(150 MHz,CDCl3):δ=19.3,52.6,63.5,125.3,127.9,129.2,130.0,130.4,136.0,150.0,163.4
MS(EI):m/z=207[M+].
2.2.5(E)-间三氟甲基苯乙酮肟的合成
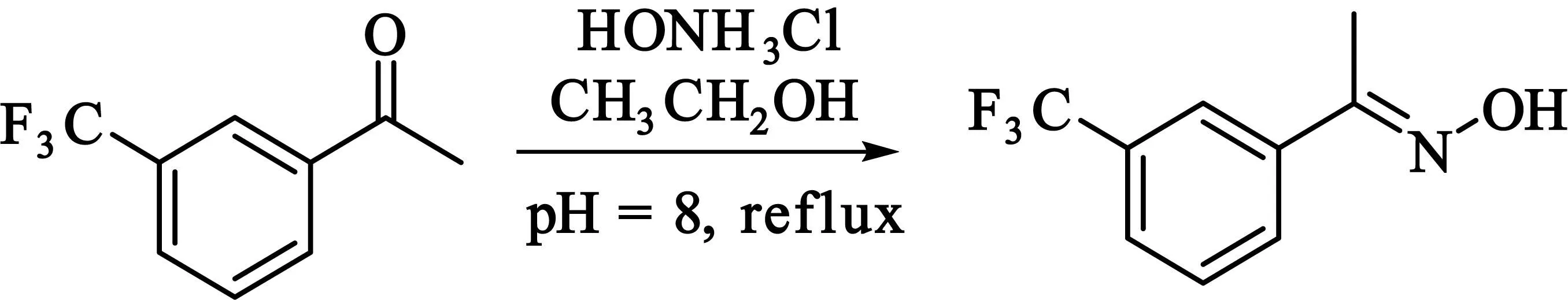
在50 mL三颈瓶中加入0.5 g(2.5 mmol)间三氟甲基苯乙酮、0.25 g(3.5 mmol)盐酸羟胺、10 mL无水乙醇,加入氢氧化钠固体,调溶液的pH在8左右,搅拌回流12 h后,倒入冰水中,用浓盐酸调pH值为2,此时溶液中有大量固体物出现,过滤,用蒸馏水洗固体至中性,抽干,得淡土黄色固体0.426 g,产率79%.
1H NMR(600 MHz,CDCl3):δ=3.88(s,3H),4.07(s,3H),4.34(s,3H),7.15(d,J=7.5 Hz,1H),7.36~7.42(m,2H),7.49(d,J=7.5 Hz,1H)
13C NMR(150 MHz,CDCl3):δ=30.6,52.9,63.7,128.3,128.6,129.7,130.1,130.4,135.7,148.9,163.0
MS(EI):m/z=285[M+].
2.2.6肟菌酯的合成

将101 mg(1.875 mmol)甲醇钠加入到10 mL DMF中,在室温下,加入300 mg(0.74 mmol)(E)-间三氟甲基苯乙酮肟,加热搅拌15 min后,冰浴下加入500 mg(0.87 mmol)(E)-2-溴甲基-α-甲氧亚胺基苯乙酸甲酯的DMF(6 mL),室温下搅拌反应,5 h停止反应,倒入冰水中,乙酸乙酯萃取,无水硫酸钠干燥,旋去乙酸乙酯,再经硅胶柱层析纯化,得到475.3 mg(E,E)-2-[1′-(3′-三氟甲基苯基)-乙基-亚胺-氧-甲苯基]-2-羰基乙酸甲酯-O-甲酮肟白色固体,产率为78%.
1H NMR(600 MHz,CDCl3):δ=1.64(s,1H),2.31(s,3H),7.52(t,J=7.8 Hz,1H),7.63(d,J=7.7 Hz,1H),7.82(d,J=7.8 Hz,1H),7.90(s,1H)
13C NMR(150 MHz,CDCl3):δ=12.0,123.0,125.3,125.8,129.0,129.2,130.9,137.3,155.0
MS(EI):m/z=203[M+].
3反应结果与讨论
3.1 酰腈水解生成酮酸
腈在酸性或者碱性条件下都可以回流水解得到羧酸,本文为了直接得到酮酯采用酸性条件水解.腈的酸性水解机理如下[10]:

氰基和羰基类似,可以质子化,质子化后的N原子很容易与水发生亲核加成,然后再消除质子得到酰胺,然后继续在酸性条件下水解得到相应的羧酸.在水解之后再酯化得到2-甲基-α-甲氧亚胺基苯乙酸甲酯,合成中主要的副产物是邻甲基苯甲酸甲酯,主要是由于在高温下2-甲基-α-甲氧亚胺基苯乙酸甲酯不稳定,容易脱去一个羰基生成2-甲基苯甲酸甲酯,所以该反应最好控制在40~50 ℃,避免副产物的生成.
3.2 N-溴代丁二酰亚胺参与的溴化反应
NBS是一种较为温和的溴代试剂,在不同条件下,可以溴化烯丙位、苄位、羰基的邻位,相对于液溴选择性更高,也可以在芳环上进行溴代,另外,还可以对烯烃进行加成.而对于苄位的溴化,温度不能过高,因为苄位相当活泼,如若温度过高会使得苄位的二溴代物的量增多[11],因此选择适当的温度对该类反应至关重要.本文基于NBS的选择性较高,而且溴化温度无需太高,所以选择NBS对苄位进行溴代.该类溴化反应一般采用一些无水的非极性惰性溶剂,如四氯化碳、苯、石油醚等,这样可以避免自由基反应以及其它一些副反应的发生.
4结语
世界农药经过几十年的探索与发展已经取得了巨大进步,其市场前景也一片大好.随着时代的进步以及环保观念的深入人心,肟菌酯具有高效、低毒、高活性以及低残留已成为当今世界农药翘楚.通过对肟菌酯传统生产工艺的研究,并在此基础上对其合成工艺进行改善,以降低生产成本,满足广大消费者的需求.
参考文献:
[1] 陆玉峰,柏亚罗.Stmbilurins类杀菌剂的作用机制与化学合成[J].现代农药,2003,2(2):29-33.
[2] Reuveni M,Can J.Activity of trifloxystrobin against powdery and downy mildew diseases of grapevines[J].Canadian Journal of Plant Pathology,2001,23(1),52-59.
[3] Bostanian N J,Larocque N.Laboratory tests to determine the intrinsic toxicity of four fungicides and two insecticides to the predacious mite Agistemus fleschneri[J].Phytoparasitica,2001,29(3):215-222.
[4] Brand S,Kardorff U.O-Benzyl oxime ethers and fungicides containing them[P].EP:463488,1993.
[5] Ziegler H,Neff D.Process for the preparation of arylacetic ester derivatives via palladium-catalyzed cross coupling reaction[P].WO:9520569,1995.
[6] Pfiffner A,Trah S,Ziegler H.Method for the production of derivatives of phenylacetic acid esters[P].EP:600835,1996.
[7] Assercq J M,Pfiffner A,Pfaff W.Process for the preparation of o-chloromethyl-phenylglyoxylic acid derivatives[P].US:5756811,1998.
[8] 李焰,周叶兵,张洪权,等.新型含氟杀菌剂trifloxystrobin的合成研究[J].华中师范大学学报:自然科学版,2005,39(1),54.
[9] 刘长令; 李正名. 新型先导化合物4-[4-(3,4-二甲氧苯基)-2-甲基噻唑-5-甲酰基]吗啉的设计、合成与生物活性[J].农药,2004,43(4):157-159.
[10] 刑其毅,裴伟伟,徐瑞秋,等.基础有机化学[M].北京:高等教育出版社,2005.
[11] 吴警,裴文. N-溴代丁二酰亚胺在有机反应中的研究进展[J].广州化工,2011,39(9),31.
(编辑崔思荣)
中图分类号:O631
文献标志码:A
文章编号:1674-358X(2015)03-0001-07
收稿日期:2015-04-20
基金项目:山东省重点研发计划项目(2015GSF119016);青岛市科技计划项目(11-2-4-3-(6)-jch)
作者简介:朱习军(1964-),男,山东菏泽人,教授,博士,硕士生导师,主要从事图像处理研究.
On the Synthesis of the Green Fungicide of Trifloxystrobin YUAN Yu, JIN Chaojun, LIU Yang
(School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002)
Abstract:Trifloxystrobin is one of the strobilurin fungicides,which has the advantages of broad-spectrum, high activity,low toxicity and so on.And the market of it is vastitude.Trifloxystrobin contains(E)-β-methoxyacrylate sbunit,and study on the mode of action of trifloxystrobin revealed that these compounds inhibit the respiration chain of fungi.This study adopted backstepping analysis to explore two synthetic routes of key intermediates of methyl 2-oxo-2-(o-tolyl)acetate,which began with the 1-(o-tolyl)ethanone and the 2-methylbenzoic acid.Then trifloxystrobin was obtained starting from methyl 2-oxo-2-(o-tolyl)acetate via reaction with methoxyamine,bromization and condensation with(E)-1-(3-(trifluoromethyl)phenyl)ethanone oxime.The total yield was 25.7%.
Key words:green fungicide; backstepping analysis; trifloxystrobin; synthesis
猜你喜欢
现代商贸工业(2016年14期)2016-12-27
安徽理工大学学报·自然科学版(2016年4期)2016-12-23
考试周刊(2016年85期)2016-11-11
农业与技术(2016年15期)2016-11-09
科技视界(2016年18期)2016-11-03
科教导刊·电子版(2016年19期)2016-08-19
科技视界(2016年15期)2016-06-30
科技视界(2016年10期)2016-04-26
科技视界(2016年9期)2016-04-26
科技视界(2016年3期)2016-02-26
