
Comparison of conventional and supported liquid extraction methods for the determination of sitagliptin and simvastatin in rat plasma by LC-ESI-MS/MS
2015-12-22 10:51RmeshMnjulBijrgiSrmSitDevi
B.Rmesh,N.Mnjul,S.R.Bijrgi,V.U.M.Srm,P.Sit Devi,✽
aNatural Products Chemistry Division,Indian Institute of Chemical Technology,Tarnaka,Hyderabad 500607,India
bMedicinal Chemistry&Pharmacology Division,Indian Institute of Chemical Technology,Tarnaka,Hyderabad 500607,India
Comparison of conventional and supported liquid extraction methods for the determination of sitagliptin and simvastatin in rat plasma by LC-ESI-MS/MS
B.Ramesha,N.Manjulaa,S.R.Bijargib,V.U.M.Sarmaa,P.Sita Devia,✽
aNatural Products Chemistry Division,Indian Institute of Chemical Technology,Tarnaka,Hyderabad 500607,India
bMedicinal Chemistry&Pharmacology Division,Indian Institute of Chemical Technology,Tarnaka,Hyderabad 500607,India
Rat plasma;
Simvastatin;
Sitagliptin;
Supported liquid
extraction
Three extraction methods were compared for their efficiency to analyze sitagliptin and simvastatin in rat plasma by LC-MS/MS,including(1)liquid-liquid extraction(LLE),(2)solid phase extraction(SPE)and(3)supported liquid extraction(SLE).Comparison of recoveries of analytes with different extraction methods revealed that SLE was the best extraction method.The detection was facilitated with ion trap-mass spectrometer by multiple reactions monitoring(MRM)in a positive ion mode with ESI.The transitions monitored were m/z 441.1→325.2 for simvastatin,408.2→235.1 for sitagliptin and 278.1→260.1 for the IS.The lower limit of quantification(LLOQ)was 0.2 ng/mL for sitagliptin and 0.1 ng/mL for simvastatin.The effective SLE offers enhanced chromatographic selectivity, thus facilitating the potential utility of the method for routine analysis of biological samples along with pharmacokinetic studies.
©2015 Xi’an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/3.0/).
1. Introduction
Sitagliptin and simvastatin combination(Fig.1)is used together with a proper diet and exercise to treat type 2 diabetes(T2D). Patients with T2D are at risk of vascular complications including cardiovascular disease[1-3],and therapy with statins in this population is widely recommended[4,5].The combination of sitagliptin,a dipeptidyl peptidase 4(DPP-4)inhibitor which improves glycemic control[6,7],and simvastatin,a well characterized lipid-lowering agent[8],may be considered an appropriate approach to management of this disease[4-9].Recently,the US Food and Drug Administration(FDA)has approved sitagliptinand simvastatin(JuvisyncW,Merck Sharp&Dohme Corp.,NJ, USA)[10],a fixed-dose combination prescription medication that contains two previously approved medications in a single tablet for use by adults who need both sitagliptin and simvastatin.Juvisync was approved in dosage strengths for sitagliptin/simvastatin of 100/10 mg,100/20 mg and 100/40 mg.
✽Corresponding author.Tel.:+91 40 27160123x1733. E-mail address:sitadeviiict@gmail.com(P.S.Devi).
Peer review under responsibility of Xi’an Jiaotong University.
2095-1779©2015 Xi’an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BYNC-ND license(http://creativecommons.org/licenses/by-nc-nd/3.0/).
Fig.1 Chemical structures of(A)sitagliptin,(B)simvastatin and(C)venlafaxine HCl(IS).
Sample preparation is an important step in the biopharmaceutical analysis process because of the difficulties that follow with complex sample matrices,e.g.plasma and urine.Sample preparation typically takes 80%of the total analysis time[11].Solid phase extraction(SPE)and liquid-liquid extraction(LLE)are among the most commonly used sample preparation methods for biological sample analysis.SPE is a time-consuming extraction for method development and sample analysis,even though it is feasible for automation.LLE requires a large volume of organic solvent that is harmful to human health and usually needs a large number of specimens.Furthermore,emulsification occurs too frequently during the extraction;thus,repeatability is not satisfactory.SLE is a promising sample cleanup technology that is particularly suitable for the 96-well format operation.Similar to the traditional LLE,SLE provides very clean extracts with a high recovery.
Several analytical methods,available in the literature,include tandem mass spectrometry method,liquid chromatography and gas chromatography-mass spectrometry(GC-MS)methods for the determination of sitagliptin alone or in combination with other drugs in biological samples[12-20].Other methods are also available for the determination of simvastatin in other drugs in biological samples or in their individual form[21-32].However, Burugula et al.[33]developed an LC-MS/MS for the analysis of sitagliptin and simvastatin in human plasma,which employed LLE with a total run time of 3.0 min for each sample.
The present communication detailed our attempts to develop and validate a reliable bioanalytical method for simultaneous determination of sitagliptin and simvastatin in rat plasma by ion trap LC-MS/ MS using SLE.A direct comparison of SLE with traditional extraction techniques,namely LLE and SPE,will demonstrate the preserved analytical advantages of this new method,with the benefit of increased sample throughput.SLE is similar to LLE but differs from it in that the sample is totally absorbed onto a solid phase, containing a modified form of diatomaceous earth,on which the extraction occurs.The retention time of sitagliptin,simvastatin and the IS was as low as 0.81,1.31 and 1.62 min,facilitating a short run time of 2.0 min.Improved experimental conditions facilitated a rapid,simple and sensitive LC-MS/MS method for simultaneous determination of sitagliptin and simvastatin in rat plasma,with a very characteristic separation pattern and enhanced chromatographic selectivity.The potential utility of the method has been thoroughly enhanced,where subtle but distinguishable separations with very low analytical run times were accomplished with Oyster ODS3 column.Herein,we present the experimental conditions and the application of the same for pharmacokinetic studies and bioanalytical evaluation.
2. Experimental
2.1. Materials and reagents
ce samples of sitagliptin(purity>99.30%)and simvastatin(purity>99.82%)were kind gift from Sipra labs(Hyderabad, India)whereasvenlafaxinehydrochloride(purity>99.25)was obtained from Micro Labs Ltd.(Bangalore,India).Water used for the LC-MS/MS analysis was prepared using a Synergy Milli Q water purification system.HPLC-grade acetonitrile,methanol and acetic acid,formic acid and methyl tert-butyl ether(MTBE)were purchased from Merck Ltd.(Mumbai,India).Ammonium acetate and ammonium formate were supplied by Qualigens Pvt.Ltd.(Bangalore, India).Sample tubes were obtained from Tarsons Pvt.Ltd.(Kolkata, India).For SLE extraction,SLE+,1 mL supported liquid extraction columns(Biotage,Uppsala,Sweden)were used.Freshly obtained drug-free rat plasma was collected from male Wistar rats in our laboratory and stored at-80°C prior to use.
2.2. LC-MS/MS analysis
Chromatographic separation was performed on an Oyster ODS3 (4.6 mm×50 mm,3 μm)column using the isocratic mode of elution.LC-MS/MS studies were performed using an Agilent 1100 MSD ion-trap-SL mass spectrometer with electrospray ionization(ESI)source in positive ion mode equipped with a degasser (G1379A),binary pump(G1312A),autosampler(G1329A),autosampler thermostat(G1329B)and diode array detector(G1315B). The data were acquired and processed using Chemstation 5.3 (Agilent Technologies,Waldbronn,Germany).An isocratic elution with acetonitrile:5 mM ammonium acetate buffer(pH 4.5±0.05) (85:15,v/v)as mobile phase was pumped at a flow rate of 0.75 mL/min;the sample injection volume was 20 μL with column temperature maintained at ambient conditions.At a time when most of the contemporary work from other laboratories was carried out using either a single or triple quadruple instruments,in the present work,an ion trap mass spectrometric analysis was deliberately employed because this mode also facilitated excellent analytically quantitative and mass selective resolutions[34]even for compounds structurally unrelated to diverse physicochemical properties.The sensitivity of the multiple reaction monitoring (MRM)was optimized by testing with an infusion of 0.1 μg/mL each of analytes and IS in mobile phase.Detection of the ions was
carried out in MRM mode,by monitoring the transition pairs of m/ z 408.2-235.1 for sitagliptin,m/z 441.1-325.2 for simvastatin and m/z 278.1-260.1 for the IS.Nitrogen was employed as the nebulizer and curtain gas.Collision-induced dissociation was achieved using helium as collision gas.The ion source conditions were set as follows:temperature,335°C;nebulizer gas,35 psi;dry gas,10.0 L/min;skimmer,40.0 V;capillary exit,128.0 V;trap drive,44.5;maximum accumulation time,200 ms;and Icc target, 20,000.
2.3. Preparation of standard and quality control(QC)samples Calibration standards were prepared by spiking the working standard solution into a pool of drug-free rat plasma in order to obtain the following concentrations:0.2,0.4,5.0,30,60,120,200, 275,350 and 450 ng/mL of sitagliptin and 0.1,0.2,5.0,10,20,50, 100,250,350 and 450 ng/mL of simvastatin.The concentrations of QC samples of sitagliptin and simvastatin were as follows:0.3 (quality control sample at low concentration,LQC),150(quality control sample at medium concentration,MQC),and 400(quality control sample at high concentration,HQC)ng/mL and 0.15 (LQC),150(MQC)and 400(HQC)ng/mL in blank plasma.The spiked samples were then treated as described in sample preparation.All the samples were stored at-80°C.
2.4. Sample preparation methods
2.4.1. LLE
LLE was carried out by the procedure described by Burugula et al. [33].A 200 μL aliquot of rat plasma sample was mixed with 50 μL of the IS working solution(50 ng/mL).To this,100 mL of 100 mM ammonium acetate buffer(pH 4.5±0.05)was added. After overtaxing for 15 s,a 4 mL aliquot of the extraction solvent (MTBE and n-hexane,70:30,v/v)was added and the sample was vortexed for 30 s and then centrifuged for 5 min at 4000 rpm.The organic layer was transferred to a 10 mL glass test tube and evaporated at 40°C under a stream of nitrogen.The dried extract was reconstituted in 250 μL of the mobile phase and transferred into auto-injector vials and 20 μL aliquot was injected into the chromatographic system.
2.4.2. SPE
A 200 μL aliquot of rat plasma sample was mixed with 50 μL of the working solution of IS(50 ng/mL)followed by 500 μL of water,and the contents were mixed by vortex for 10 s and subjected to SPE.The sample mixture was loaded onto a StrataX 33 μm polymeric sorbent cartridge(100 mg/3 mL)that was preconditioned with 2.0 mL of methanol followed by 2.0 mL Milli Q water.Washing of cartridges was performed with 2 mL of 20% methanol in water.
Analytes and IS were eluted with 2.0 mL of the mobile phase. An aliquot of 20 μL of the sample was injected into the LC-MS/ MS system for analysis.
2.4.3. SLE
A 200 μL aliquot of sample was diluted with 50 μL of the IS solution(50 ng/mL)followed by 300 μL of 5 mM ammonium acetate buffer(pH 4.5±0.05)and then mixed for 10 s and loaded onto an Isolute SLE+cartridge.A minimum positive pressure was applied to facilitate the sample absorption into the cartridge in less than 10 s.After the analytes were allowed to equilibrate with the sorbent for a minimum of 5 min,the compounds were eluted with an aliquot of 1 mL of extraction solvent.The extraction solvent was eluted by applying a little positive pressure from the top with a rubber bulb for fast elution and evaporated to dryness under a stream of heated nitrogen at 40°C.Finally the residues were reconstituted in 400 μL methanol solution(aq)and vortexed at medium speed for approximately 1 min.
A 20 μL aliquot of the reconstituted solution was injected into the LC-MS/MS system for analysis.
3. Results and discussion
3.1. Method development
To develop a sensitive and reliable LC-ESI-MS/MS method,it is important to optimize the chromatographic and mass spectrometric conditions,as well as to obtain an efficient and simple extraction procedure for all the analytes along with a suitable IS.Choosing the appropriate IS is an important aspect to achieve acceptable method performance,especially with LCMS/MS,where matrix effects can lead to poor analytical results. In the present method,several compounds were investigated to find a suitable IS,and finally venlafaxine hydrochloride was chosen because it offers high recovery,less analytical run time and does not hamper the employed analytical evaluation at any stage of analysis.
3.2. Optimization of chromatography
In the present work,chromatography was performed on several reversed-phase columns like Agilent Zorbax C8(4.6 mm×150 mm,5 μm),Xterra RP C18(4.6 mm×250 mm,5 μm),XDB-C18(4.6 mm×150 mm,5 μm),Oyster C8(4.6 mm×50 mm,5 μm), OysterODS3 C18(4.6 mm×50 mm,3 μm),AtlantisdC18(4.6 mm×150 mm,5 μm),and SUPELCO Discovery C8(4.6 mm×250 mm,5 μm)to achieve a short run time,symmetric peak shapes,minimum matrix interference and solvent consumption.This was investigated by appropriate changes to mobile phase composition(aqueous and organic part),buffer pH and flow rate. Thus to arrive at an ideal solvent system,we attempted various combinations of methanol/acetonitrile along with buffers(ammonium formate/formic acid,ammonium acetate/acetic acid,ammonium acetate/ammonia)having different ionic strengths(1-10 mM)in the pH range of 3.0-8.0 and volume ratios(60:40, 70:30,80:20,85:15 and 90:10,v/v).Further,the effect of flow rate was also studied from 0.6 to 1.2 mL/min,which was always considered to be responsible for acceptable chromatographic resolution.Use of a 5 mM ammonium acetate buffer(pH 4.5±0.05)helped in achieving a good response for MS detection in the positive ionization mode for both the analytes and the IS.It was found that a mixture of acetonitrile and 5 mM ammonium acetate buffer(pH 4.5±0.05;85:15,v/v)could achieve this purpose and was finally employed as the suitable mobile phase. An Oyster ODS3 column facilitated well peak shape and response even at the lowest concentration level for both the analytes and IS (Fig.2E).The mobile phase was operated at a flow rate of 0.75 mL/min.The retention time of sitagliptin,simvastatin and the IS was low enough to be 0.81,1.31 and 1.62 min,respectively, allowing a short run time of 2.0 min.
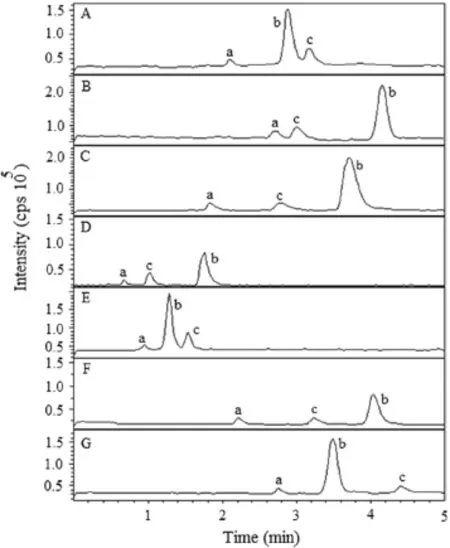
Fig. 2 Typical chromatograms of analytes (a=sitagliptin, b=simvastatin and c=IS)acquired at different columns to evaluate peak resolution.(A):Agilent Zorbax C8(4.6 mm×150 mm,5 μm); (B):Xterra RP C18(4.6 mm×250 mm,5 μm);(C):XDB-C18(4.6 mm×150 mm,5 μm);(D):OysterC8(4.6 mm×50 mm, 5 μm);(E):Oyster ODS3 C18(4.6 mm×50 mm,3 μm);(F):Atlantis dC18(4.6 mm×150 mm,5 μm);and(G):SUPELCO Discovery C8(4.6 mm×250 mm,5 μm).
3.3. Optimization of mass spectrometry
As both the analytes have very different structures and ionization behavior,the tuning of mass parameters was carried out using ESI in positive.Standard solutions of the analytes and IS were directly infused along with the mobile phase into the mass spectrometer with ESI as the ionization source.LC-MRM is a very powerful technique for pharmacokinetic studies since it efficiently offers greater sensitivity and selectivity for any analytical method employed and thus the MRM technique was chosen for the assay development.The MRM parameters were optimized to maximize the response for the analyte.The most sensitive mass transition was monitored from m/z 408.2 to 235.1 for sitagliptin,from m/z 441.1 to 325.2 for simvastatin and from m/z 278.1 to 260.1 for the IS.The product ions of these compounds are shown in Fig.3.
3.4. Optimization of sample preparation
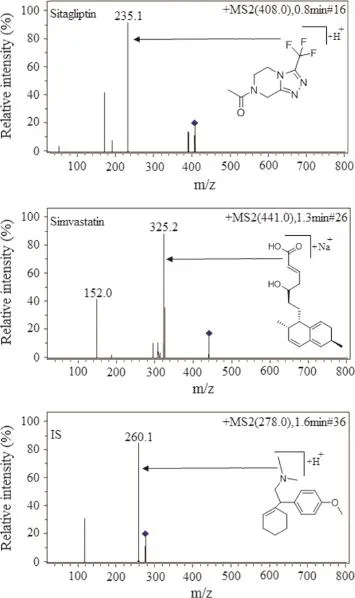
Fig.3 Full scan product ion of precursor ions of sitagliptin, simvastatin and IS.
In our studies,different extraction methods such as LLE,SPE and SLE were studied and compared.LLE with mixture of MTBE and n-hexane(70:30,vv),SPE with StrataX 33 μm polymeric sorbent cartridge(100 mg/3 mL)and SLE with Isolute SLE+cartridge were investigated during the method development.As shown in Fig.4A,the extraction efficiencies for sitagliptin and simvastatin were improved significantly with SLE.Therefore,SLE was employed to treat the sample.The parameters evaluated in method development were extraction solvent,recovery of analytes after evaporation and reconstitution of the samples,and plasma volume to be extracted.For sample elution,a number of organic solvents were investigated for SLE experiments.These were ethyl acetate, diethyl ether,MTBE,iso-propanol and dichloromethane.Finally, ethyl acetate was found to give cleaner extract and higher recovery (Fig.4B).The recoveries of analytes were studied after evaporation and reconstitution in aqueous methanol solutions of various strengths(90%,80%and 70%).Finally,a solution consisting of 90%methanol(aq)was chosen for reconstitution of the samples. Here,a combination of 200,300,400 and 500 μL diluent and 200, 300,400 and 500 μL of plasma was also investigated.We found that 200 μL plasma and 400 μL diluent improved the extraction recovery.
3.5. Method validation
The validation process was carried out according to Guidance for Industry-Bioanalytical Method Validation recommended by U.S. Food and Drug Administration[35].The method was validated for
selectivity,sensitivity(LLOQ),linearity,precision,accuracy, recovery,matrix effect,carryover and stability.
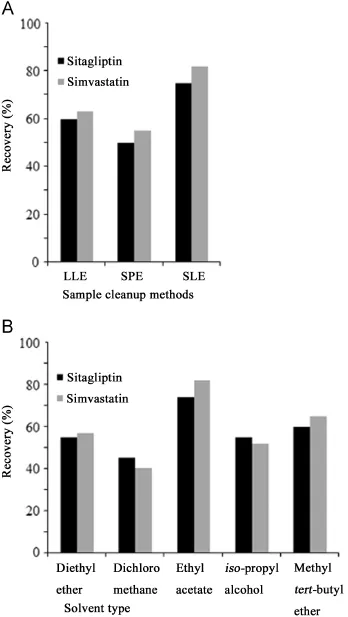
Fig.4 (A)Comparison of sample cleanup methods and (B) comparison of various extraction solvents.
3.5.1. Selectivity
Selectivity of the method was evaluated by analyzing six different blank plasma samples to investigate the potential interferences at the LC retention time for the analytes and IS. The selectivity of the method was tested by comparing the chromatograms of blank plasma and the spiked plasma.Under the above conditions,the retention time of sitagliptin,simvastatin and IS was 0.81,1.31 and 1.62 min,respectively.All plasma lots were found to be free of interferences with the compounds of interest.As shown in Fig.5,there was no interference at the retention time of sitagliptin,simvastatin and IS.
3.5.2. Calibration curves
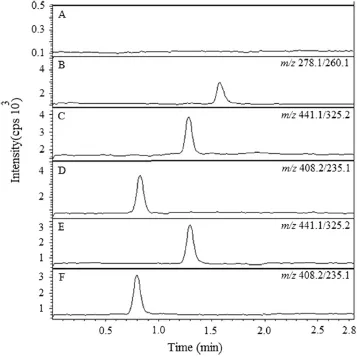
Fig.5 Representative MRM ion-chromatograms of(A)blank plasma and(B)blank plasma spiked with IS at 50 ng/mL,(C)simvastatin at 0.05 ng/mL(LLOQ),(D)sitagliptin at 0.1 ng/mL (LLOQ),(E) simvastatin and(F)sitagliptin in rat plasma sample collected at 120 min after oral administration.
To prepare the standard curves,appropriate amounts of sitagliptin and simvastatin were added to blank plasma yielding the following concentrations:0.1,0.2,0.4,5.0,30,60, 120,200,275,350 and 450 ng/mL for sitagliptin and 0.05,0.1, 0.2,5.0,10,20,50,100,250,350 and 450 ng/mL for simvastatin.These samples were then prepared in triplicate according to the procedure described above.The calibration curve was found to be linear over the concentration range of 0.10-450 ng/mL for sitagliptin and 0.05-450 ng/mL for simvastatin.The calibration curve was constructed by plotting the peak area ratio of analyte to IS vs the nominal concentration in plasma.The data were subjected to statistical analysis using a linear regression model,showing that the regression equation was y=0.002x+0.047,with correlation coefficient(R2)greater than 0.996 for sitagliptin and y=0.044x+0.112,with correlation coefficient(R2)greater than 0.998 for simvastatin,where y represents the peak area ratio of analytes to that of IS and x represents the concentration of analytes in ng/mL.
3.5.3. Precision and accuracy
The intra-day assay precision and accuracy were obtained by analyzing six replicates of LLOQ and QC samples on a single day. The inter-day assay precision and accuracy were obtained by analyzing six replicates of LLOQ and QC samples on 3 different days.Intra-day and inter-day precisions of the method were expressed by[standard deviation/mean concentration]×100%. Accuracy of the method was expressed by[(mean measured concentration-nominal concentration)/nominal concentration]×100%. The relative percentage error(RE%)of the mean value should be within±15%at each concentration except for the LLOQ,where the RE should be within±20%.The precision was required to be less than 20%at the LLOQ level and less than 15%at other concentrations. As shown in Table 1,the values for both intra-and inter-day accuracy and precision were found to be within the acceptable criteria.
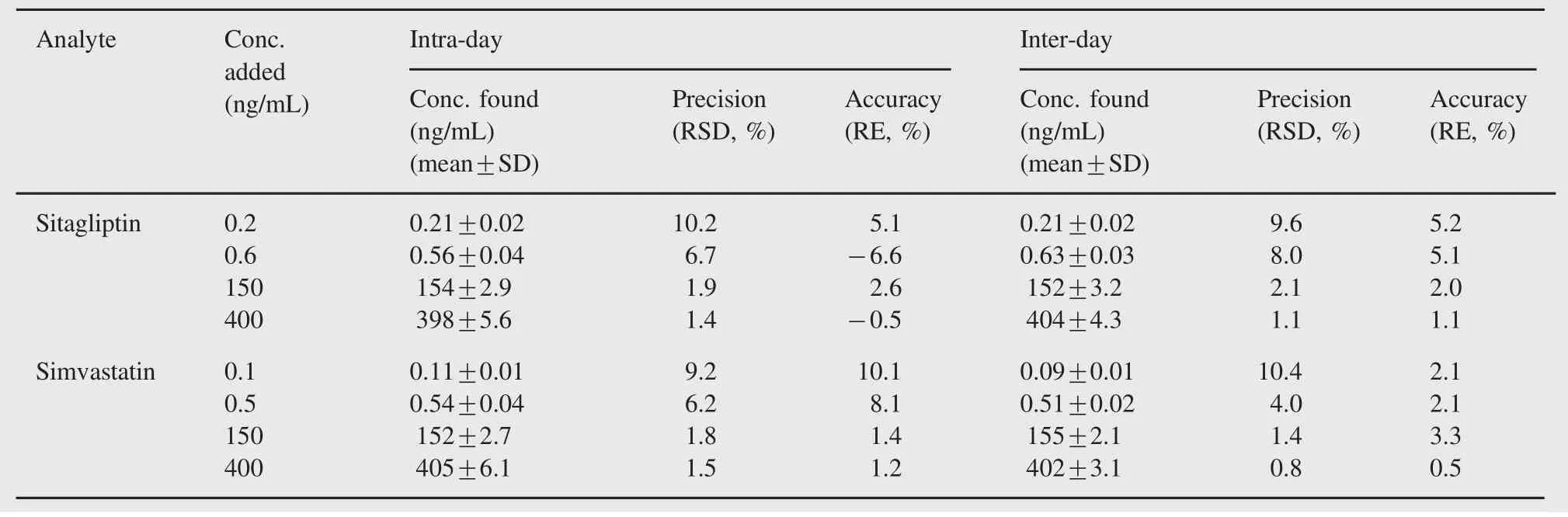
Table 1 Intra-and inter-day precision and accuracy for the detection of sitagliptin and simvastatin in rat plasma(n=6).
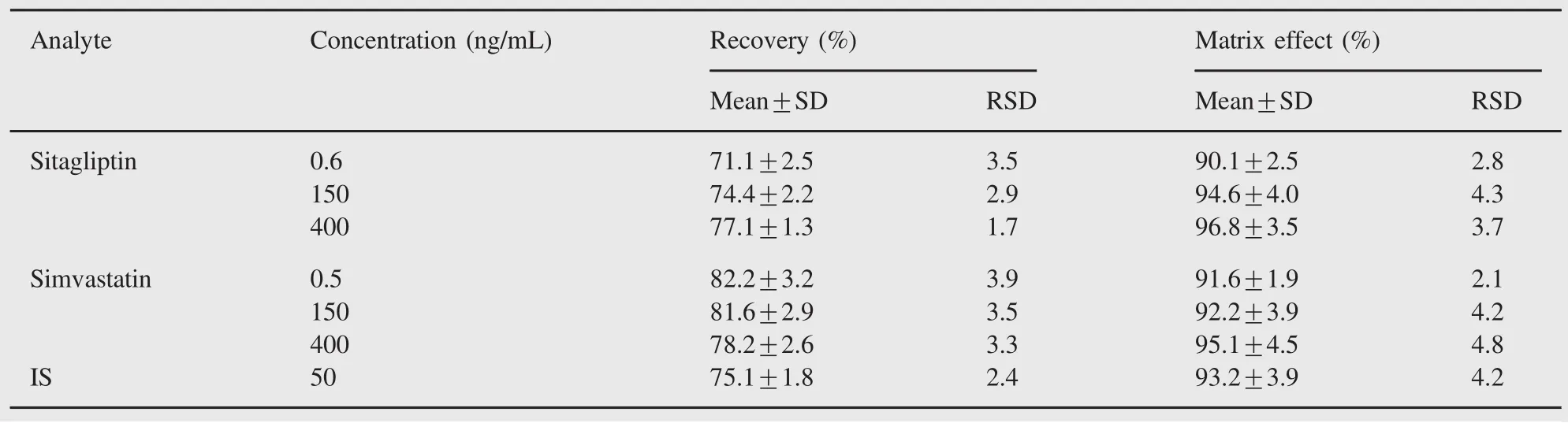
Table 2 The recovery and matrix effect of sitagliptin,simvastatin and IS(n=6).
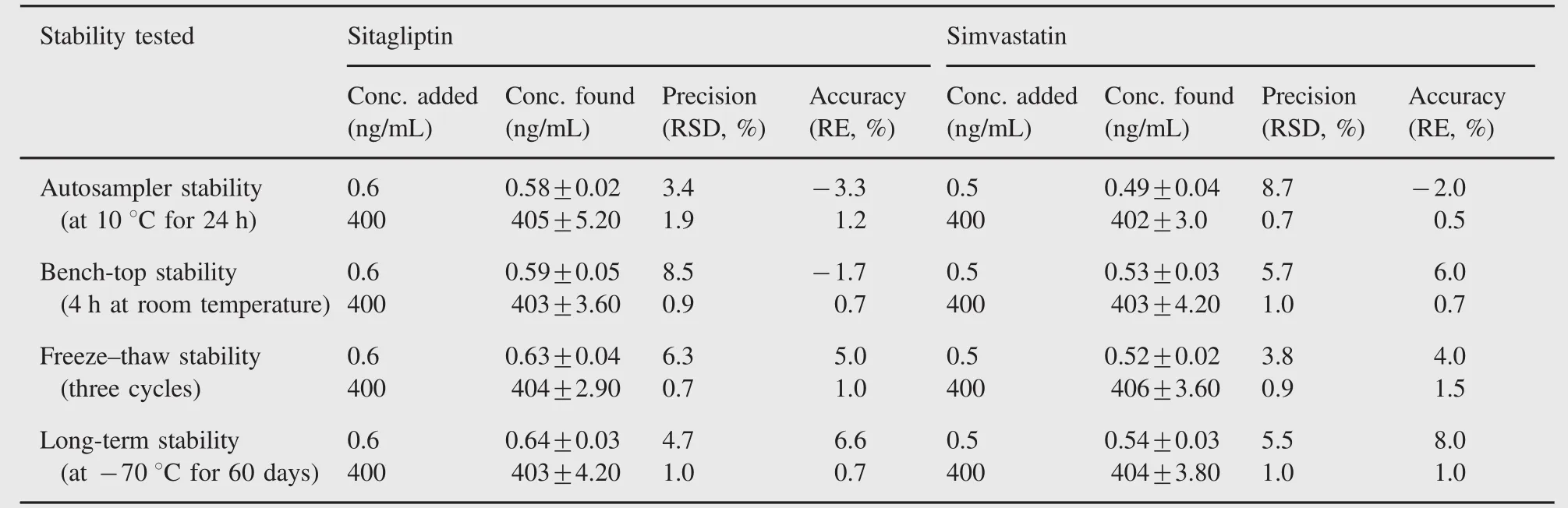
Table 3 Stability of sitagliptin and simvastatin in rat plasma(n=6).
3.5.4. Recovery and matrix effect
The extraction recovery for each analyte for three levels of QC samples was assessed by comparing the peak areas for extracted spiked plasma samples with the peak areas for pure compounds of the same concentrations in solvent.The recovery of the IS was evaluated at the concentration used in sample analysis.Matrix effect was investigated to ensure precision,selectivity and sensitivity that were not compromised by the matrix screened. Blank biological samples were extracted and then spiked with the analytes at three QC levels and IS in five replicates.The corresponding peak areas were compared with those of standard solutions,and the peak area ratio was defined as the matrix effect. Table 2 shows the results of the recovery and the matrix effect for sitagliptin,simvastatin and IS.The results indicated satisfactory recovery and no significant relative matrix effect,which could negatively influence quantitation results.
3.5.5. Carryover
The carryover was evaluated by analyzing a blank sample immediately after the upper limit of quantification(ULOQ)sample of the standard curve.The carryover level should be<20%of the response observed for the analyte at LLOQ and<5%of the response observed for the IS at the working concentration.No peak was observed at the retention time of any analyte or IS in the chromatogram of a blank sample analyzed after the injection of ULOQ sample,indicating the absence of carryover.
3.5.6. Stability
The stability tests of the analytes were designed to cover expected conditions concerning the handling of clinical samples.The stabilities of sitagliptin and simvastatin in plasma at different concentrations were examined under different study conditions,i.e.keeping at room temperature for 4 h(bench-top stability)and storing at-70°C for at least 2 months(long-term stability).The stabilities of sitagliptin and simvastatin in plasma extracts were also tested by keeping the samples at 10°C for 24 h(autosampler stability).Freeze/thaw stability was determined after freezing(-30°C)and thawing for three cycles.All the stability studies were conducted at LQC and HQC levels using six replicates at each level.Samples were considered to be stable if assay values were within the acceptable limits of accuracy(≤15%RE)and precision(≤15%RSD).The results are summarized in Table 3.The results indicated that sitagliptin and simvastatin were stable for the entire period of the experiment.
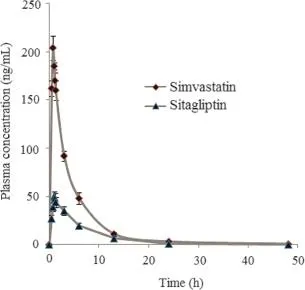
Fig.6 Mean plasma concentration-time profiles of sitagliptin and simvastatin after administration to rats(n=5).
3.6. Application of the method to a pharmacokinetic study
The applicability of the developed bioanalytical method(SLE-LCESI/MS)for sitagliptin and simvastatin in rat plasma was demonstrated by the results obtained from pharmacokinetic studies conducted in three male Wistar rats weighing 250±10 g approximately,which were fasted overnight before and 4 h after dosing.(The study was approved by the Animal Ethical Committee of Indian Institute of Chemical Technology,Hyderabad.)Each rat received an oral dose of 15 mg/kg sitagliptin and 25 mg/kg of simvastatin in gum acacia suspension. Blood samples were collected from orbital sinus into EDTA coated tubes at 0,0.5,0.75,1,1.25,1.50,3,6,12,24 and 48 h time intervals after drug administration.The blood samples were centrifuged at 5000 rpm for 10 min and the plasma samples were stored at-80°C until analysis.The samples were determined by the chromatographic conditions described as in Section 2.2.Plasma drug concentration-time data were subjected to noncompartmental pharmacokinetic analysis using linear trapezoidal rule.Fig.6 shows the mean plasma concentration-time profile of sitagliptin and simvastatin.The pharmacokinetic parameters such as Cmax,Tmax,t1/2,AUC0-t,AUC0-∞and MRT for sitagliptin and simvastatin are summarized in Table 4.
4. Conclusion
A simple,rapid and sensitive LC-MS/MS method was developed and validated for the simultaneous determination of sitagliptin and simvastatin in rat plasma with simple SLE method in a very short run time of 2 min.The present SLE method has the highest recovery compared with other extraction with techniques.The validation study successfully evaluated intra-day and inter-day precision,selectivity,sensitivity,linearity,recovery,carryover, matrix effect and sample stability.The results of the rat plasma validation parameters were well within the acceptable limits. Further,the plasma matrix components do not interfere with the analysis.The LLOQ was 0.2 ng/mL for sitagliptin and 0.1 ng/mL for simvastatin,using 100 μL of plasma sample.The method offers significant advantages over those previously reported,in terms of improved sensitivity,requiring a small volume of extraction solvent,simplicity of extraction procedure and short overall analytical run time.The efficiency of SLE and chromatographic run time of 2.0 min per sample render the method useful in high-throughput bioanalysis.The fully validated method is simple, highly sensitive,specific,robust,and has been successfully applied to pharmacokinetic study in rats.This method is suitable for routine analysis of a large number of biological samples.

Table 4 Main pharmacokinetic parameters of sitagliptin and simvastatin in rat plasma(n=6).
Acknowledgments
The authors are grateful to Dr.M.Laxmikantam,Director,CSIRIICT,for providing facilities to perform this work.
References
[1]J.Stamler,O.Vaccaro,J.D.Neaton,et al.,Diabetes,other risk factors, and 12-yr cardiovascular mortality for men screened in the multiple risk factor intervention trial,Diabetes Care 16(1993)434-444.
[2]M.J.Garcia,P.M.McNamara,T.Gordon,et al.,Morbidity and mortality in diabetics in the Framingham population.Sixteen year follow-up study,Diabetes 23(1974)105-111.
[3]I.M.Stratton,A.I.Adler,H.A.Neil,et al.,Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes(UKPDS 35):prospective observational study,Br.Med.J. 321(2000)405-412.
[4]American Diabetes Association,Standards of medical care in diabetes,Diabetes Care 34(2011)S11-S61.
[5]L.Ryden,E.Standl,M.Bartnik,et al.,Guidelines on diabetes,prediabetes,and cardiovascular diseases:executive summary.The task force on diabetes and cardiovascular diseases of the European society of cardiology(ESC)and of the European association for the study of diabetes(EASD),Eur.Heart J.28(2007)88-136.
[6]A.Karasik,P.Aschner,H.Katzeff,et al.,Sitagliptin,a DPP-4 inhibitor for the treatment of patients with type 2 diabetes:a review of recent clinical trials,Curr.Med.Res.Opin.24(2008)489-496.
[7]N.A.Thornberry,A.E.Weber,Discovery of JANUVIA(Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes,Curr.Top.Med.Chem.7(2007)557-568.
[8]T.R.Pedersen,J.A.Tobert,Simvastatin:a review,Expert Opin. Pharmacother.5(2004)2583-2596.
[9]P.M.Kearney.,L.Blackwell,R.Collins,et al.,Efficacy of cholesterollowering therapy in 18,686 people with diabetes in 14 randomised trials of statins:a meta-analysis,Lancet 371(2008)117-125.
[10]U.S.Food and Drug Administration,October,2011,available at:〈http://www.fda.gov/〉.
[11]G.Vas,K.Vekey,Solid-phase microextraction:a powerful sample preparation tool prior to mass spectrometric analysis,J.Mass Spectrom.39(2004)233-254.
[12]E.Uçakturk,Development of a gas chromatography-mass spectrometry method for the analysis of sitagliptin in human urine,J.Pharm. Biomed.Anal.74(2013)71-76.
[13]G.Vishal,R.Rajeshwari,D.Ranjeet Prasad,et al.,Simultaneous quantification of aliskire,valsartan and sitagliptin by LC with fluorescence detection:evidence of pharmacokinetic interaction in rats,Chromatographia 76(2013)515-521.
[14]W.Zeng,D.G.Musson,A.L.Fisher,et al.,Determination of sitagliptin in human urine and hemodialysate using turbulent flow online extraction and tandem mass spectrometry,J.Pharm.Biomed. Anal.46(2008)534-542.
[15]W.Zeng,D.G.Musson,A.L.Fisher,et al.,Determination of MK-0431 in human plasma using high turbulence liquid chromatography online extraction and tandem mass spectrometry,Rapid Commun. Mass Spectrom.20(2006)1169-1175.
[16]R.Nirogi,V.Kandikere,K.Mudigonda,et al.,Sensitive liquid chromatography tandem mass spectrometry method for the quantification of sitagliptin,a DPP-4 inhibitor,in human plasma using liquidliquid extraction,Biomed.Chromatogr.22(2008)214-222.
[17]C.Hess,F.Musshoff,B.Madea,et al.,Simultaneous identification and validated quantification of 11 oral hypoglycaemic drugs in plasma by electrospray ionisation liquid chromatography-mass spectrometry,Anal.Bioanal.Chem.400(2011)33-41.
[18]W.Zeng,Y.Xu,M.Constanzer,et al.,Determination of sitagliptin in human plasma using protein precipitation and tandem mass spectrometry,J.Chromatogr.B 878(2010)1817-1823.
[19]R.N.Rao,P.K.Maurya,S.Khalid,et al.,Development of a molecularly imprinted polymer for selective extraction followed by liquid chromatographic determination of sitagliptin in rat plasma and urine,Talanta 85(2011)950-957.
[20]J.G.Swales,R.T.Gallagher,M.Denn,et al.,Simultaneous quantitation of metformin and sitagliptin from mouse and human dried blood spots using laser diode thermal desorption tandem mass spectrometry, J.Pharm.Biomed.Anal.55(2011)544-551.
[21]G.Carlucci,P.Mazzeo,L.Biordi,et al.,Simultaneous determination of simvastatin and its hydroxy acid form in human plasma by high-performance liquid chromatography with UV detection, J.Pharm.Biomed.Anal.10(9)(1992)693-697.
[22]H.Ochiai,N.Uchiyama,K.Imagaki,et al.,Determination of simvastatin and its active metabolite in human plasma by columnswitching high-performance liquid chromatography with fluorescence detection after derivatization with 1-bromoacetylpyrene,J.Chromatogr.B Biomed.Sci.Appl.694(1997)211-217.
[23]L.Wang,M.Asgharnejad,Second-derivative UV spectrometric determination of simvastatin in its tablet dosage form,J.Pharm. Biomed.Anal.21(6)(2000)1243-1248.
[24]M.Jemal,Z.Ouyang,M.L.Powell,et al.,Direct-injection LC-MSMS method for high-throughput simultaneous quantitation of simvastatin and simvastatin acid in human plasma,J.Pharm.Biomed.Anal. 23(2000)323-340.
[25]J.J.Zhao,A.Y.Yang,J.D.Rogers,et al.,Effects of liquid chromatography mobile phase buffer contents on the ionization and fragmentation of analytes in liquid chromatographic/ionspray tandem mass spectrometric determination,J.Mass Spectrom.37(2002)421-433.
[26]H.Yang,Y.Feng,Y.Luan,et al.,Determination of Simvastatin in human plasma by liquid chromatography-mass spectrometry,J. Chromatogr.B 785(2003)369-375.
[27]N.Zhang,A.Yang,J.D.Rogers,et al.,Quantitative analysis of simvastatin and its β-hydroxy acid in human plasma using automated liquid-liquid extraction based on 96-well plate format and liquid chromatography-tandem mass spectrometry,J.Pharm.Biomed.Anal. 34(2004)175-187.
[28]A.Y.Yang,L.Sun,D.G.Musson,et al.,Application of a novel ultralow elution volume 96-well solid-phase extraction method to the LC/MS/MS determination of simvastatin and simvastatin acid in human plasma,J.Pharm.Biomed.Anal.38(2005)521-527.
[29]B.Barrett,J.Huclova,V.Bořek-Dohalský,et al.,Validated HPLCMS/MS method for simultaneous determination of simvastatin and simvastatin hydroxy acid in human plasma,J.Pharm.Biomed.Anal. 41(2006)517-526.
[30]N.Lucie,V.Hana,S.Dalibor,et al.,Ultra high performance liquid chromatography tandem mass spectrometric detection in clinical analysis of simvastatin and atorvastatin,J.Chromatogr.B 877 (2009)2093-2103.
[31]N.Sultana,M.Saeed Arayne,N.Shafi,et al.,Development of a RP-HPLC method for the simultaneous analysis of diltiazem and statin:application in pharmaceuticals and human serum,Anal. Methods 2(2010)1571-1576.
[32]A.M.Saeed,S.Najma,M.A.Zeeshan,et al.,High-performance liquid chromatographic analysis of pioglitazone,gliquidone,rosuvastatin and simvastatin in formulations and human serum,Chin.J.Chem.28 (2010)1998-2002.
[33]L.Burugula,R.Mullangi,N.R.Pilli,et al.,Simultaneous determination of sitagliptin and simvastatin in human plasma by LC-MS/MS and its application to a human pharmacokinetic study,Biomed. Chromatogr.27(2013)80-87.
[34]R.K.Jonscher,J.R Yates,The quadrupole ion trap mass spectrometer-a small solution to a big challenge,Anal.Biochem.244(1997)1-15.
[35]US Department of Health and Human Services,Food and Drug Administration,Center for Drug Evaluation and Research,Guidance for Industry,Bioanalytical Method Validation,May 2001,available at:〈http://www.fda.gov/cder/guidance〉.
Received 12 May 2014;revised 2 November 2014;accepted 20 November 2014 Available online 2 December 2014
 Journal of Pharmaceutical Analysis2015年3期
Journal of Pharmaceutical Analysis2015年3期
- Journal of Pharmaceutical Analysis的其它文章
- Non-covalent binding analysis of sulfamethoxazole to human serum albumin:Fluorescence spectroscopy,UV-vis,FT-IR,voltammetric and molecular modeling
- Determination of diclofenac in pharmaceutical preparations by voltammetry and gas chromatography methods
- Quality evaluation of synthetic quorum sensing peptides used in R&D
- Extraction,characterization and biological studies of phytochemicals from Mammea suriga
- Liquid chromatography-tandem mass spectrometry method for the estimation of adefovir in human plasma:Application to a pharmacokinetic study
- Selective extraction of dimethoate from cucumber samples by use of molecularly imprinted microspheres