
Quality evaluation of synthetic quorum sensing peptides used in R&D
2015-12-22 10:51FrederickVerbekeEvelienWynendaeleSarahBraetMatthiasHondtBartDeSpiegeleer
Frederick Verbeke,Evelien Wynendaele,Sarah Braet, Matthias D’Hondt,Bart De Spiegeleer
Drug Quality and Registration(DruQuaR)Group,Faculty of Pharmaceutical Sciences,Ghent University, Ottergemsesteenweg 460,B-9000 Ghent,Belgium
Received 3 October 2014;revised 23 November 2014;accepted 9 December 2014 Available online 31 January 2015
Quality evaluation of synthetic quorum sensing peptides used in R&D
Frederick Verbeke1,Evelien Wynendaele1,Sarah Braet, Matthias D’Hondt,Bart De Spiegeleer✽
Drug Quality and Registration(DruQuaR)Group,Faculty of Pharmaceutical Sciences,Ghent University, Ottergemsesteenweg 460,B-9000 Ghent,Belgium
Received 3 October 2014;revised 23 November 2014;accepted 9 December 2014 Available online 31 January 2015
Quorum sensing peptides; Quality;
Peptides are becoming an important class of molecules in the pharmaceutical field. Closely related peptide-impurities in peptides are inherent to the synthesis approach and have demonstrated to potentially mask biomedical experimental results.Quorum sensing peptides are attracting high interest in R&D and therefore a representative set of quorum sensing peptides,with a requested purity of at least 95.0%,was evaluated for their purity and nature of related impurities. In-house quality control(QC)revealed a large discrepancy between the purity levels as stated on the supplier’s certificate of analysis and our QC results.By using our QC analysis flowchart,we demonstrated that only 44.0%of the peptides met the required purity.The main compound of one sample was even found to have a different structure compared to the desired peptide.We also found that the majority of the related impurities were lacking amino acid(s)in the desired peptide sequence.Relying on the certificates of analysis as provided by the supplier might have serious consequences for peptide research,and peptide-researchers should implement and maintain a thorough in-house QC.
©2015 Xi’an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1. Introduction
Peptides are becoming an important class of molecules in the biomedical and pharmaceutical fields owing to their high affinity, strong selectivity for their targets and low toxicity[1].Despite potential limitations such as overall low oral bio-availability,low metabolic resistance,potential immunogenicity,poor membrane permeability and financial aspects,several peptide drugs have entered the market[2,3].The promising future of peptide therapeutics is further highlighted by the number of peptides in clinical and preclinical phases[4].In 2012,approximately 200 peptides entered the clinical phase,while another 400 were at advanced preclinical stages[4-6].
Quorum sensing peptides are a group of peptides currently attracting high interest.Quorum sensing,the process of cell-tocell communication between bacteria,has been the subject of a great number of scientific research papers[7-9].Three main groups of quorum sensing molecules can be distinguished:N-acylhomoserine lactone derivatives(AHL or auto-inducer-1), quorum sensing peptides and boron-furan derivatives(autoinducer-2).The quorum sensing peptides,mainly found in Gram-positive bacteria,show a large structural diversity:short, linear fragments as well as cyclic(thiolacton)derivatives,with orwithoutpost-translationalisoprenylmodifications,are observed[10].These peptides bind to(i)bacterial membrane associated receptors,or(ii)cytoplasmatic receptors,after which the transcription of the target genes is activated.Interfering with this bacterial quorum sensing pathway might open interesting application perspectives.
Chemical peptide synthesis for medicinal purposes has become economically viable[11].The possibility to produce small, medium(5-20 residues)to large(20-50 residues)peptides has evolved dramatically,hereby frequently outperforming the biotechnological approaches as they are known to date[6].In 1965, Merrifield cleared the way for Solid-Phase Peptide Synthesis (SPPS),thus introducing and facilitating a totally new concept in peptide synthesis[12]:a peptidic chain,fixed to a solid support,is created by the consecutive addition of the appropriate amino acids. The reaction can be automated and possible solubilization-issues are avoided due to the fixation of the peptide to the solid matrix [13].During SPPS,different side-reactions can occur,resulting in several types of peptide impurities[14]:e.g.(i)diketopiperazine structures[15,16],(ii)aspartimide residues[17-19],(iii)cysteine racemization[16],(iv)diastereoisomeric products[20,21],(v) dimers[22],(vi)acid precursors and protected sequences[20], (vii)oxidation and reduction of amino acids[19,23],(viii)amino acid deletions[15,23-26],(ix)amino acid insertions[23,24],(x) products of side chain reactivity and(xi)amino acid modifications during cleavage[16,19,23,24,26-30].Thus,the crude peptides obtained from SPPS mostly will contain many by-products.These impurities,if present even after purification,have to be quantified and characterized to meet pharmaceutical regulatory requirements,but they need to be under control as well during the unregulated biomedical research and discovery phases[31,32].Due to budgetary constraints,peptides used in research are often purchased at undefined or low purity levels,e.g.70.0%[31].Generally,the peptide purity used for biomedical research and discovery purposes ranges from as low as 50.0%to more than 95.0%[22,26].However,closely related impurities of the target peptide may possess stronger binding affinities compared to the native peptide[26],thus potentially causing erroneous conclusions.Investigations by de Beukelaar et al.[33]indicated different immune responses of a protein-spanning peptide pool of 70.0%pure peptides due to the impurities.False-positive results in a HIV-vaccine trial were also ascribed to impurities in the peptide-mixtures[34]. Zhang et al.[35]were unable to reproduce their initial obestatin results in vitro,possibly due to impurities present in the initially examined peptide.Additionally,Verbeken et al.[31]observed different biofunctional responses in a set of tissue-organ bath experiments caused by impurities of the examined peptides.Impurities are thus able to potentially mask biomedical experimental outcomes and may cause false negative or positive results.
Quorum sensing peptides are currently being actively investigated,i.e.for their possible role in the crosstalk between the microbiome and its host[36,37].Therefore,a thorough in-house QC analysis of a selected set of quorum sensing peptides will be conducted,followed by identification of the observed impurities. These data not only demonstrate the need for routine QC in peptide research,but also can help in building a global overview of expected related impurities in peptides.
2. Materials and methods
2.1. Chemicals and reagents
A set of 98 representative peptides was selected from the Quorumpeps database[38].Linear peptide synthesis was conducted by an international supplier,while another supplier synthesized the cyclic peptides,both by means of Fmoc-SPPS. A minimal purity of 95.0%was requested at order.The sequences of the 98 peptides are provided in Table 1.Acetonitrile of HPLCMS and UPLC-MS grades were purchased from Fisher Scientific (Aalst,Belgium).Formic acid(LC-MS grade)and DMSO(p.a.≥99.9%)were obtained from Sigma-Aldrich(Diegem,Belgium). Trifluoroacetic acid(TFA),NaH2PO4⋅H20 and Na2HPO4werepurchased from Merck(Overijse,Belgium).Water was purified using an Arium 611 purification system(Sartorius,Gottingen, Germany)yielding≥18.2 MΩ cm quality water.Eppendorf Protein LoBind 2.0 and 1.5 mL tubes were purchased from Eppendorf (Nijmegen,the Netherlands).UPLC/HPLC vials and inserts were purchased from Waters(Milford,MA,USA).The Vydac Everest C18(250 mm×4.6 mm,5 μm)column and Alltech Prevail Organic Acid column(250 mm×4.6 mm,5 μm),both protected with guard columns,were obtained from Grace(Deerfield,IL, USA).The Acquity UPLC BEH300 C18column(100 mm×2.1 mm,1.7 μm)with guard column was purchased from Waters (Milford,MA,USA).
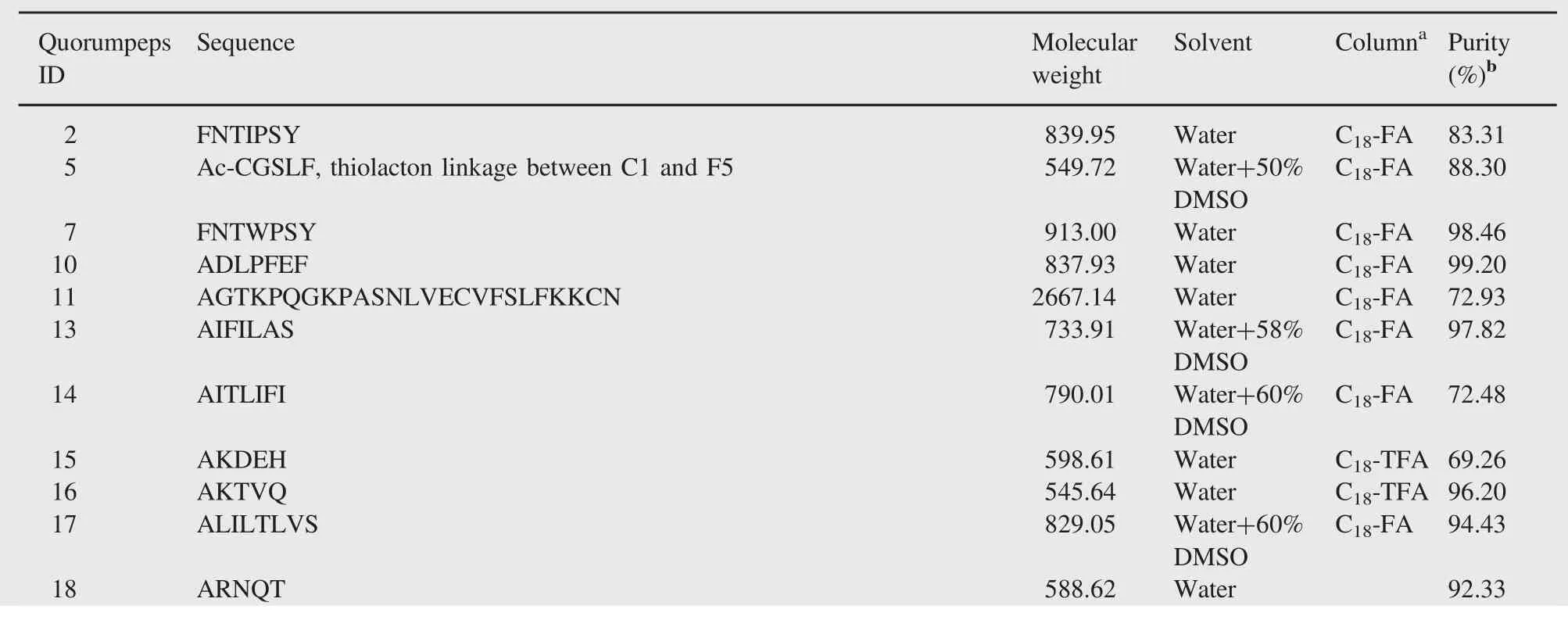
Table 1 Peptides information.
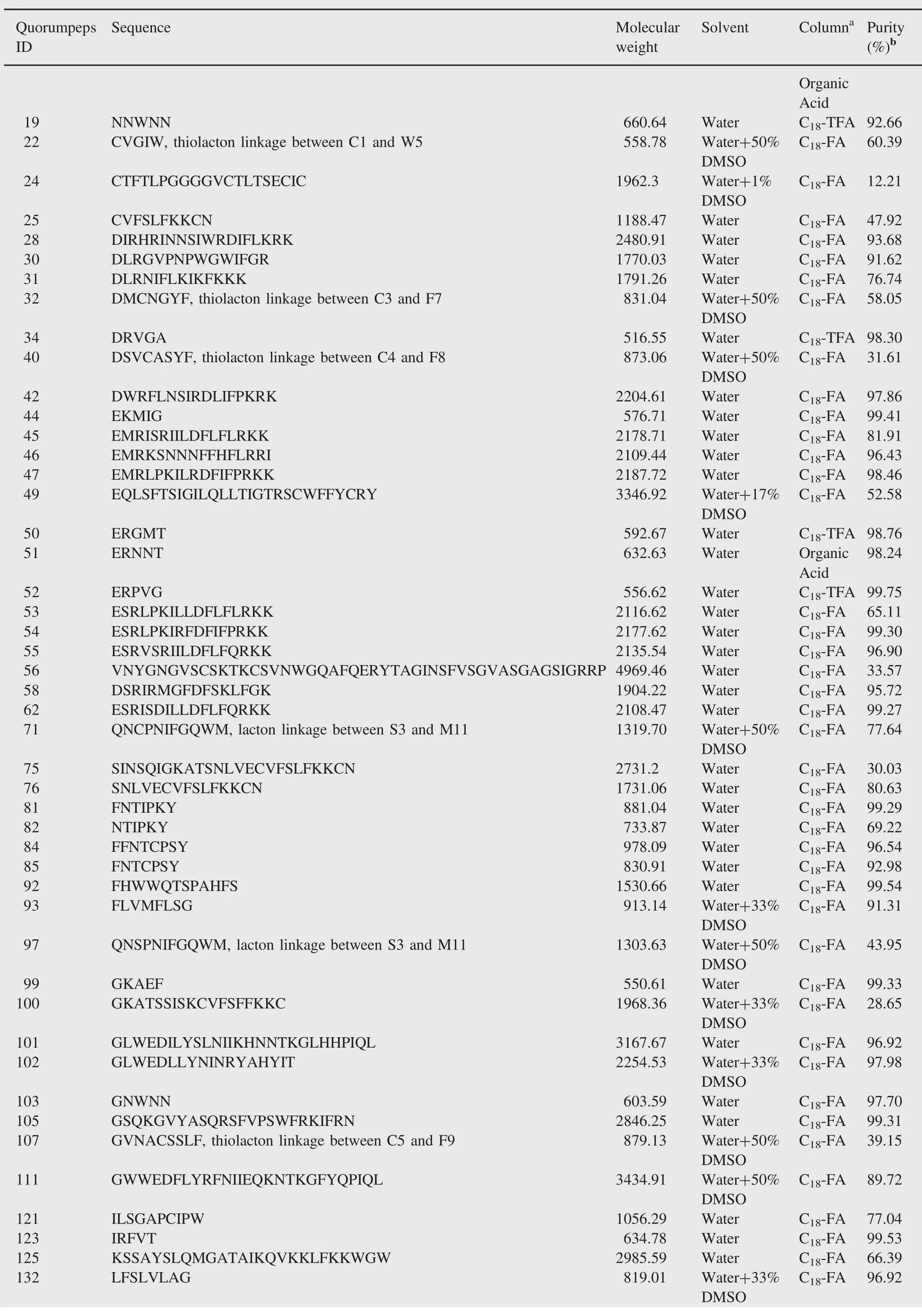
Table 1(continued)
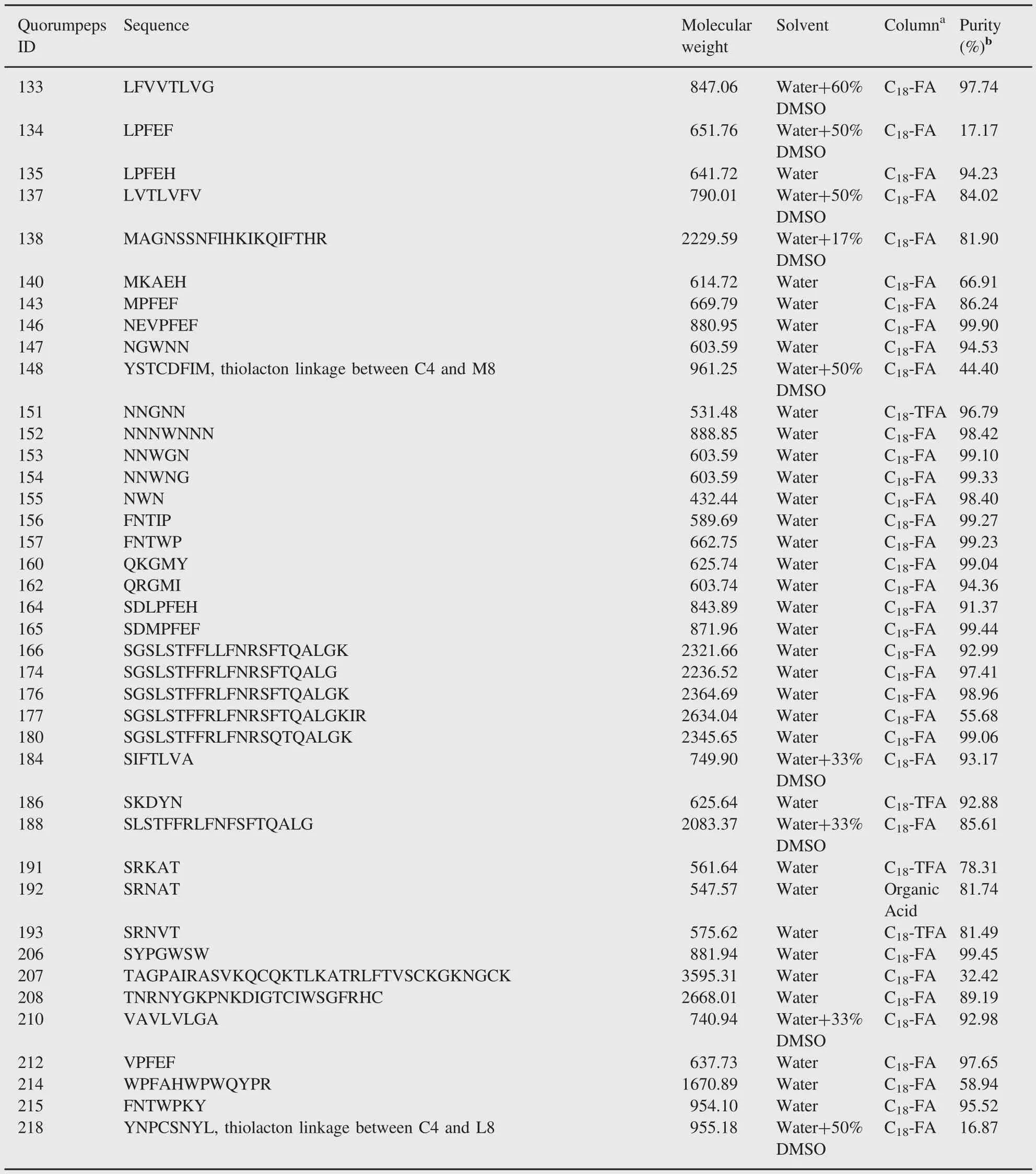
Table 1(continued)
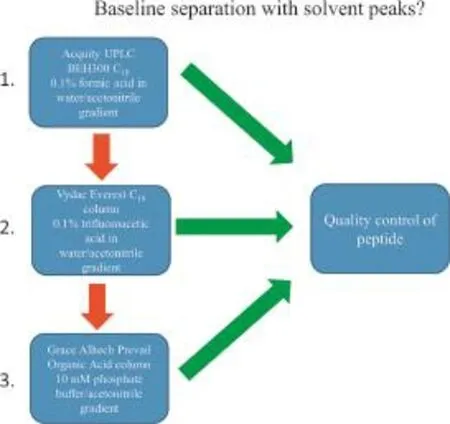
Fig.1 (U)HPLC column flow chart for quorum sensing peptides analysis.

Fig.2 Purity levels of quorum sensing peptides determined by inhouse QC.
2.2. Sample preparation
0.5-1 mg of each lyophilized,linear peptide was dissolved in water or a mixture of water and DMSO,depending on the calculated log P values and the solubility in water,to obtain a concentration of approximately 1.0 mg/mL(Table 1).The quantity of DMSO used was determined experimentally,by adding DMSO stepwise to the peptide-water suspension until no more precipitate was observed.The dissolved peptides were stored at-35°C.The cyclic peptides(i.e.ID5,22,32,40,71,97,107,148 and 218) were dissolved in DMSO-water(50/50,V/V)to obtain a concentration of approximately 0.5 mg/mL.
2.3. HPLC and UPLC
2.3.1. QC purity profiling
Three LC systems were consecutively applied in the analysis.To avoid carry-over peaks,50%acetonitrile in water was always used as needle wash.
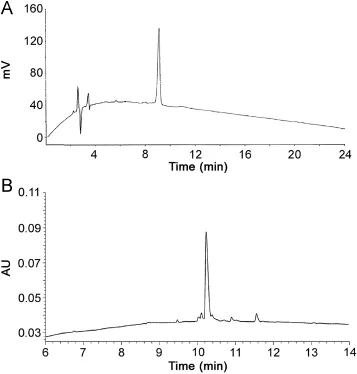
Fig.3 The in-house QC control shows a distinct difference in peptide purity.(A)Chromatogram of supplier;Kromasil C18-5(250 mm×4.6 mm,5 μm),quantification at 220 nm;(B)chromatogram of in-house QC; Vydac Everest C18column(250 mm×4.6 mm,5 μm),quantification at 210 nm.
Each peptide was first analyzed with the Acquity UPLC BEH300 C18column.Sample compartment was kept at 5°C(± 2°C)and column was maintained at 30°C(±2°C).Injection volume was set at 2 μL for the linear and 5 μL for the cyclic peptides.Mobile phases consisted of 0.1%(m/V)formic acid in water(A)and 0.1%(m/V) formic acid in acetonitrile(B).A general linear gradient was applied running from 95%A to 20%A during 22 min,followed by returning to initial condition and re-equilibration with a flow rate set at 0.5 mL/ min.Analysis was conducted using a Waters Acquity UPLC Class BioQuaternary Solvent Manager,a Waters Acquity Bio-sample Manager,combined with a Flow Through Needle and a Waters Acquity UPLC PDA(500 nL to 10 mm path length analytical flow cell)detector(Waters,Milford,MA,USA).Empower™2 software was employed for data acquisition and analysis.Peptides and related impurities were quantified by area-normalization at 210 nm,using a sampling rate of 20 points/s and a detector time constant of 0.1 s.A reporting threshold(i.e.0.1%of main peak AUC in the UV chromatogram)and identification threshold(i.e.0.5%of main peak AUC in the UV chromatogram)as stated in the European Pharmacopoeia were applied[39].Solvent and system peaks,observed in the blank(i.e.the solvent used to dissolve a certain peptide,Table 1),were excluded.
If no retention of the peptides was observed using the Acquity UPLC BEH300 C18column,peptides were analyzed with the Vydac Everest C18column on a Waters Alliance 2695 HPLC apparatus equipped with a Waters 2695 Separations Module,combined with a Flow Through Needle,and a Waters 2996 Photodiode Array Detector with Empower™ 2 software for data acquisition.Mobile phases consisted of 0.1%(m/V)TFA in water(A)and 0.1%(m/V)TFA in acetonitrile(B).A general linear gradient was applied running from 95%A to 20%A during 30 min,followed by returning to initial condition and re-equilibration with a flow rate set at 1.0 mL/min.For peptide ID191,a slightly modified linear gradient was applied,i.e. running from 98%A to 80%A during 10 min,followed by returning to initial conditions and re-equilibration with a flow rate set at 1.0 mL/ min.Sample compartment was kept at 5°C(±2°C)and column was maintained at 30°C(±2°C);20 μL of the samples dissolved in suitable solvent composition were injected.UV detection was performed at 215 nm using a sample rate of 1.0 point/s combined
with a detector time constant of 0.2 s.The flow rate was set to 1.0 mL/min.The peptide purity was quantified through normalization by calculating the area(UV)of the most abundant peak as a percentage of total peak areas.
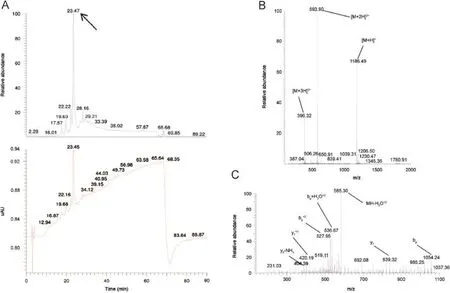
Fig.4 TIC and UV chromatogram(A),MS1(B)and MS2(C)spectra of the related impurity C1VFSLFKKC9N(disulfide bond between C1and C9)of peptide ID25 at a retention time of 23.47 min with appointed isotopic mass patterns and b-and y-fragmentation are allocated.

Fig.5 TIC and UV chromatogram(A),MS1(B)and MS2(C)spectra of the related impurity LPFE(methyl)F of peptide ID134 at a retention time of 29.75 min with appointed isotopic mass patterns and b-and y-fragment allocation.
Finally,peptides ID18,51 and 192 were analyzed using a Grace Alltech Prevail Organic Acid column(250 mm×4.6 mm,5 μm), using the same Waters Alliance 2695 HPLC equipment as used with the Vydac column.Mobile phases consisted of 10 mM phosphate buffer(pH 7)(A)and acetonitrile(B).A general linear gradient was applied running from 95%A to 60%A during 20 min,followed by returning to initial condition and reequilibration with a flow rate set at 1.0 mL/min.Sample compartment was kept at 5°C(±2°C)and column maintained at 30°C (±2°C).20 μL of the samples dissolved in suitable solvent composition were injected.UV detection was performed at 215 nm,using a sample rate of 1.0 point/s combined with a detector time constant of 0.2 s.The peptide purity was quantified through normalization by calculating the area(UV)of the most abundant peak as a percentage of total peak areas.
2.3.2. Identification of main LC peak and related impurities
Massspectroscopic characterization wasperformed with a Thermo HPLC system consisting of a Waters 2487 dual λ absorbance UV/VIS-detector(UV at 215 nm),a Finnigan LCQ ion trap mass spectrometer with electrospray ionization(ESI)and Xcalibur™software version 2.0 for data acquisition.The LC-MS equipment is yearly qualified using a tune solution and qualification solution according to the EDQM guideline[40].The tune solution consists of 100 μL 1.0 mg/mL caffeine(Sigma-Aldrich, Diegem,Belgium)in methanol,15 μL 5 nmol/μL MRFA(Met-Arg-Phe-Ala)(Research Plus Inc.,Atlantic City,NJ,USA)in 50/ 50 methanol/water(V/V),2.5 mL 0.1%(V/V)Ultramark 1621 (Alfa Aesar,Karlsruhe,Germany)in acetonitrile,50 μL glacial acetic acid(Riedel de Haen,Diegem,Belgium)and 2.34 mL of 50/ 50 methanol/water(V/V).The EDQM qualification solution consists of a 10.0 μg/mL reserpine(Flandria,Ghent,Belgium) solution in acidified methanol(i.e.1%acetic acid in methanol(V/ V)).The reserpine solution is infused in the mass spectrometer with a flow rate of 3 μL/min with a collision energy of 35.00 eV. The m/z value of 609.4 is selected as parent ion and the m/z range for the daughter ions is set to m/z 165.0-800.0.The obtained m/z values are compared with the acceptance criteria as set by the EDQM.
For the analysis of the quorum sensing peptides,column temperature was set at 30°C(±2°C)and samples were kept at room temperature.20 μL of aqueous peptide solution was injected into the HPLC system and a flow rate of 1.0 mL/min was applied. All 15 selected quorum sensing peptides were analyzed using the Vydac Everest C18column(250 mm×4.6 mm,5 μm).Mobile phases consisted of 0.1%(m/V)formic acid in water(A)and 0.1%(m/V) formic acid in acetonitrile(B)with a flow rate of 1.00 mL/min.An isocratic period consisting of 95%A during 5 min was maintained, followed by a linear gradient running from 95%A to 40%A in 60 min and returning to the initial conditions and re-equilibration afterwards. The ion transfer capillary was operated at 250°C,nitrogen was used as sheath gas(80 arbitrary units corresponding to 1.2 L/min)and helium
as auxiliary gas(20 arbitrary units corresponding to 0.2 L/min).The spray voltage was 4.50 kV and the capillary voltage was 46 V.MS1(100-2000 m/z)and MS2(data dependent,i.e.highest abundant MS1m/z value for MS2)data were obtained.All m/z values possessing an abundance higher than 25%in the according MS1spectrum were subject to identification via their respective MS2fragmentation pattern. Theoretical MS2fragmentation patterns of the peptides were calculated in silico with ProteinProspector[41](University of California,San Francisco,CA,USA).The proposed sequences of the related impurities were confirmed using the ProteinProspector generated MS2spectra. The nomenclature originally proposed by Roepstorff and Fohlman[42] was used to annotate MS2fragments.
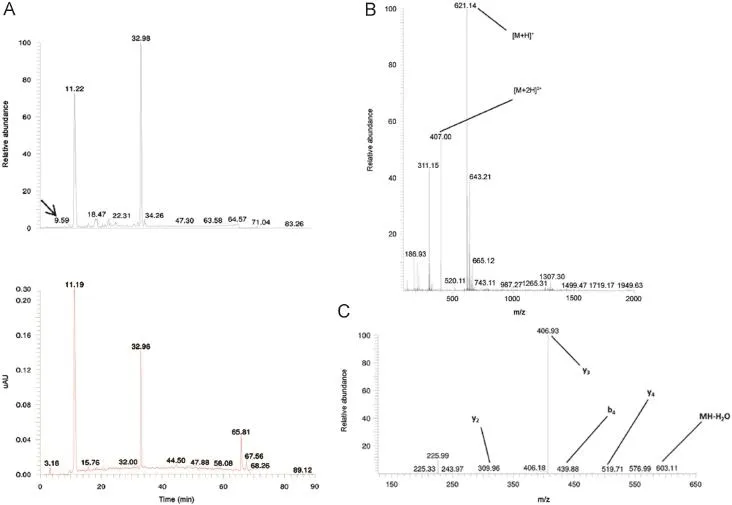
Fig.6 TIC and UV chromatogram(A),MS1(B)and MS2(C)spectra of the related impurity TIPKY of peptide ID82 at a retention time of 9.59 min with appointed isotopic mass patterns and b-and y-fragment allocation.
3. Results and discussion
3.1. Quorum sensing peptide analysis flow chart
To determine the purity of the quorum sensing peptides,we developed a UPLC/HPLC-analysis flow chart(Fig.1).Each peptide was analyzed first with the Acquity UPLC BEH300 C18column,using the method described above.A C18column with a 0.1%(m/V)formic acid in water/acetonitrile gradient is widely used for peptide analysis and was therefore selected as the initial method of analysis.If no retention of the peptides was observed, the peptideswere analyzed with the Vydac EverestC18column with TFA as ion-pairing reagent,which is known to have a narrowing effect on peak width and an increased retention. Finally,if no retention was observed with the aforementioned columns,the Prevail Organic Acid column(mobile phase of pH 7) with a 10 mM phosphate buffered water/acetonitrile gradient was used for peptide quality control.Previous experience indicated that peptides which had no retention using standard C18peptide columns might have appropriate retention using a polar-embedded column for highly hydrophilic compounds,like the Prevail Organic Acid column.
3.2. QC purity assay
Despite a requested purity of at least 95.0%,only 43 out of 98 peptides met these purity specifications(Fig.2).However,all the 98 peptides were accompanied by a certificate of analysis stating at least 95.0%purity.Our QC analysis upon arrival showed purity levels sometimes far below the demanded purity level(Table 1).In Fig.3,the chromatogram of ID76,as provided by the supplier on the certificate of analysis versus our QC analysis,is given.The mobile phases of the supplier consisted of 0.1%(m/V)TFA in acetonitrile(A)and 0.1%(m/V)TFA in water(B).A general linear gradient was applied running from 22%A to 47%A during 22 min with a Kromasil C18-5(250 mm×4.6 mm,5 μm)column with quantification at 220 nm.The peptide was analyzed in our laboratory with the Acquity UPLC BEH300 C18column,using the operational details as described above.
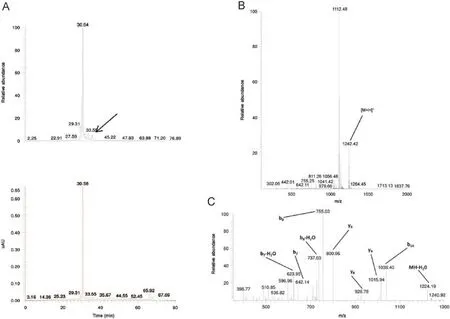
Fig.7 TIC and UV chromatogram(A),MS1(B)and MS2(C)spectra of the related impurity ILSGAPCIPWW of peptide ID121 at a retention time of 35.70 min with appointed isotopic mass patterns and b-and y-fragment allocation.
The method applied by the supplier clearly lacked selectivity to determine the purity level of the peptide.We demonstrated the necessity of a reliable QC method,i.e.the need of a suitable chromatographic system to analyze peptide purity.The supplier claimed≥95.0%purity using its system,where we determined a purity level of 80.6%.
Especially the cyclic peptides(n=9)were characterized by low purity levels:none of the peptides had a purity exceeding 90.0%.Of all cyclic peptides,1 peptide had a purity between 80.0%and 90.0%,whereas the other 8 had purity levels below 80.0%. Nevertheless,each of these peptides was accompanied by a certificate of analysis stating a purity of at least 95.0%.After communication with the suppliers,they revealed thatthey were unable to produce these cyclic peptides at the requested purity level.
3.3. Identification of related impurities
A set of 15 peptides with varying purities was selected to determine the identity of the impurities(Table 1,depicted in red).The main peak was characterized as the desired peptide for 14 out of 15 peptides.The main peak from peptide ID25 was not the requested amino acid sequence.Instead of CVFSLFKKCN,a peptide with a mass difference of-2.1 Da compared with the requested peptide sequence was noted.Therefore,the formation of a disulfide bond(and thus cyclisation)between C1and C9is suggested.Because sulfur-containing amino acids are prone to oxidation[43],a spontaneous cyclisation is suspected for this peptide(Fig.4).
The MS1spectrum of a related peptide impurity of peptide ID134(retention time of 29.75 min)showed an impurity with a monoisotopic mass of 666.22 Da(Fig.5).The difference with the monoisotopic mass of the requested peptide sequence(LPFEF) amounted to 14.04 Da,a mass difference that could be associated with the addition of a methyl group.The MS2fragmentation pattern of the related peptide impurity(Fig.5C)showed a difference of+14 Da with the requested peptide sequence fragmentation pattern at b4,y2-H2O,y4,MH-H20,while the b3fragment of requested peptide and related impurity were the same, thus suggesting the addition of a methyl group to Glu4.
A related impurity of peptide ID82(retention time 9.34 min) possessed an m/z value of 621.14(Fig.6).Compared with the desired peptide,i.e.NTIPKY,there was a mass difference of 114.02 Da,a mass difference which could be bridged by the deletion of Asn1.The suggested sequence TIPKY was confirmed by the MS2spectrum in Fig.6C.On the other hand,we also demonstrated a related impurity characterized by the incorporation of an additional amino acid at the C-terminus of peptide ID121 (retention time 35.70 min)(Fig.7).The MS2spectrum(Fig.7C) confirmed the addition of an extra Trp residue at the C-terminus of peptide ID121,thus resulting in the related impurity ILSGAPCIPWW.Another example of a related peptide impurity was the combination of both deleted and additionally inserted amino acids in the desired peptide sequence.In Fig.8,such an example is
given:ILSGAPCIPPPP is a related impurity of peptide ID121 and lacks the C-terminal Trp residue,but is characterized by 3 additional Pro residues at the C-terminus.
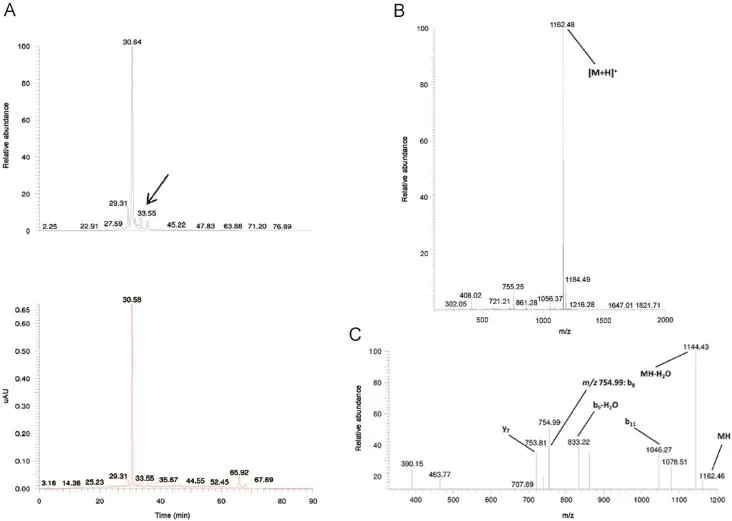
Fig.8 TIC and UV chromatogram(A),MS1(B)and MS2(C)spectra of the related impurity ILSGAPCIPPPP of peptide ID121 at a retention time of 33.55 min with appointed isotopic mass patterns and b-and y-fragment allocation.
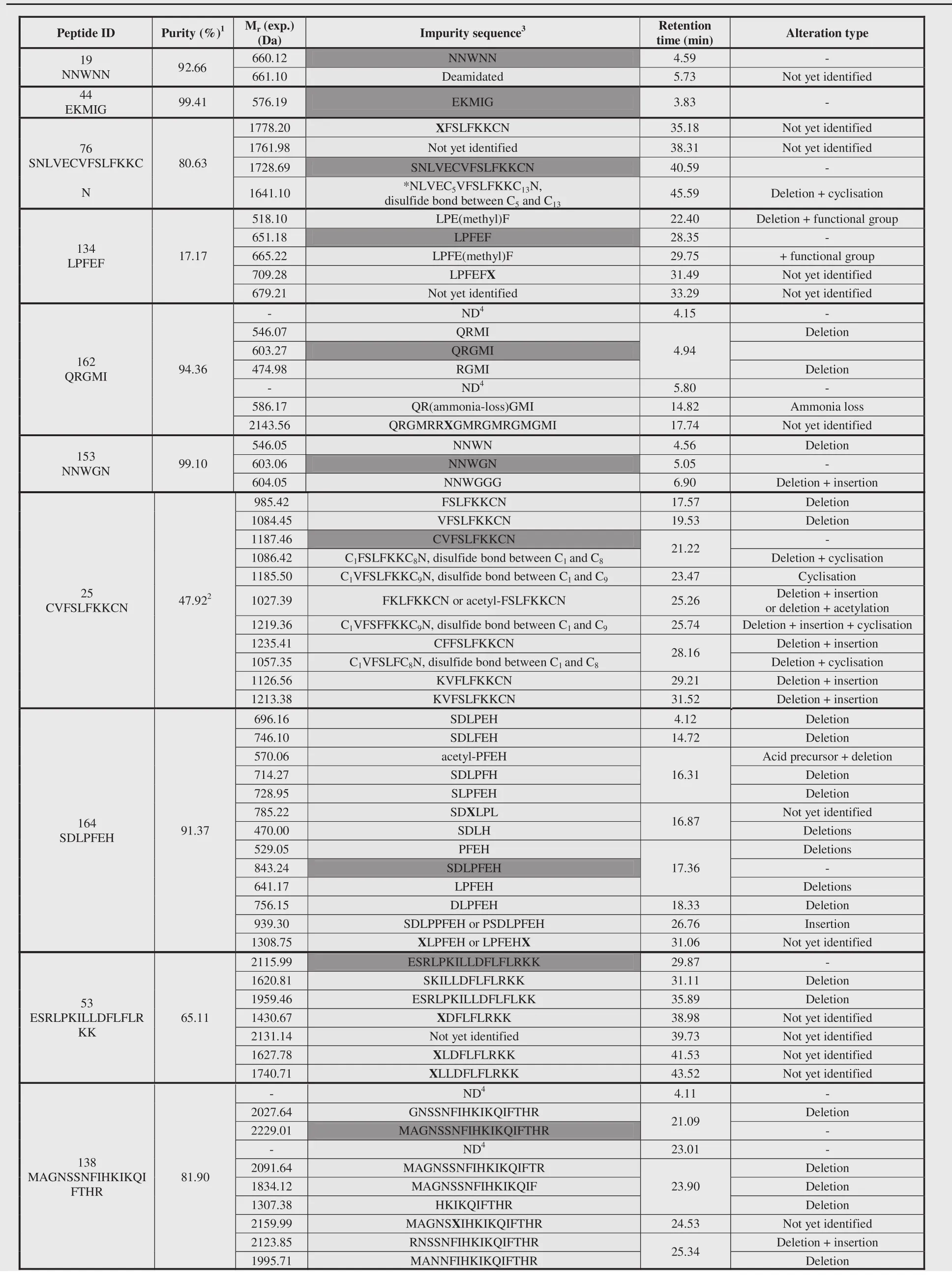
Table 2 Overview of the observed peptides(blue)and impurities(purple).
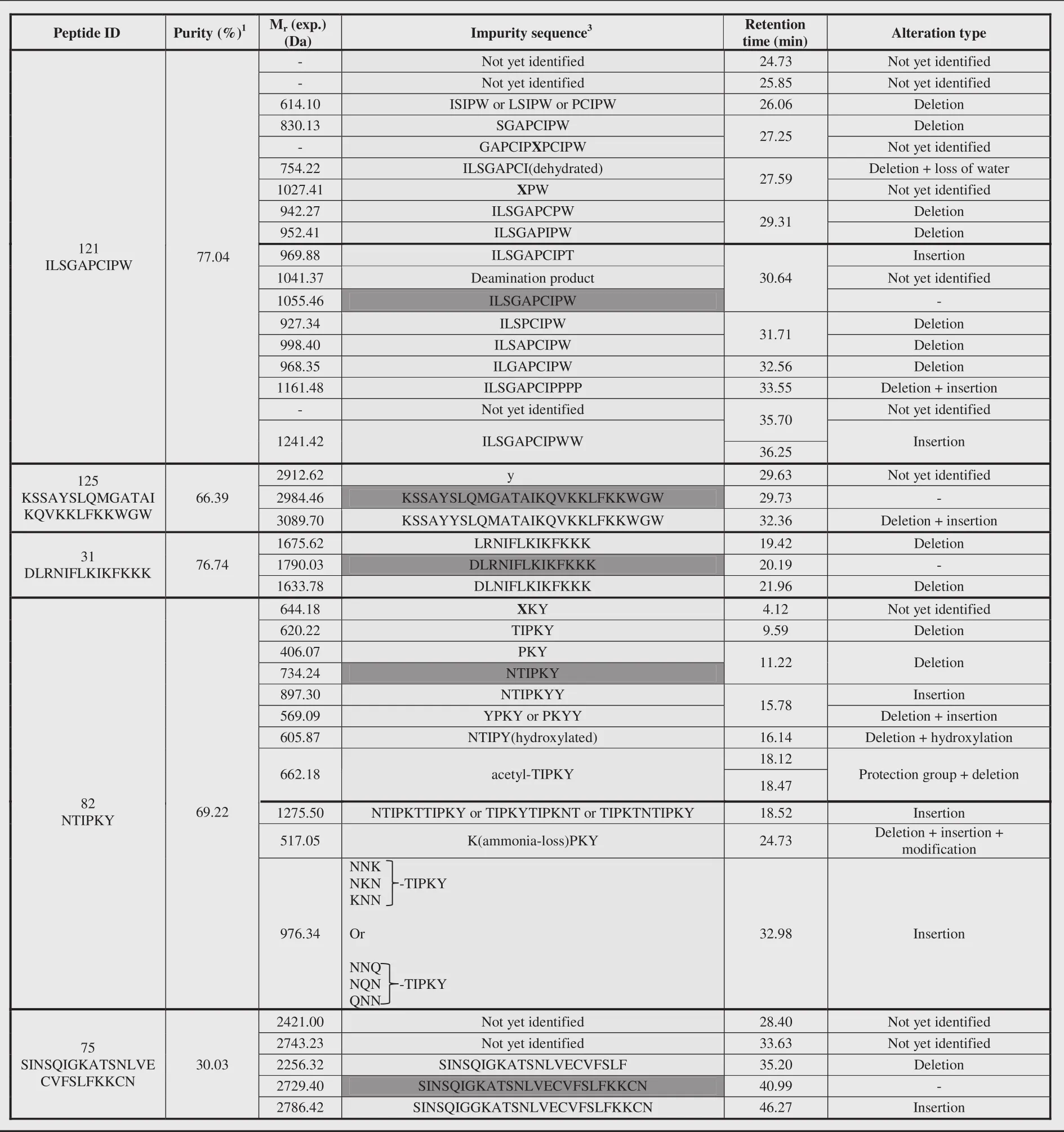
Table 2.(continued)
In total,84 impurities(Table 2)were observed and 73%could be assigned a structure related to the requested peptide by means of MS2.Analysis of these identified impurities revealed a high abundance of impurities with at least one deletion(i.e.in 74%of all identified impurities is involved)(Fig.9).Inappropriate deletion of blocking groups during synthesis results in such sequences, thereby lacking at least 1 amino acid[20,44].A minority of the observed peptide impurities were characterized by the incorporation of additional amino acids.During synthesis,an excess of amino acid equivalents used to assure a maximum coupling efficiency might result in the incorporation of additional amino acids[20,45].Impurities can also be a combination of alteration types,e.g.one or more desired amino acid(s)might be missing at the N-terminus whilst at the C-terminus additional amino acid(s) are incorporated.
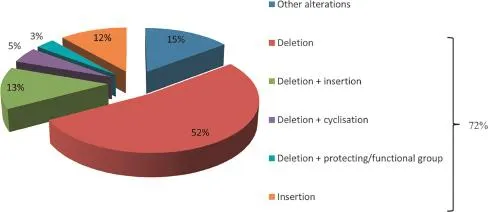
Fig.9 Types of related peptide impurities found in the 61 experimentally determined sequences.
4. Conclusions
In conclusion,“purified”synthetic peptides still contain a considerable amount and wide variety of related impurities,with peptides missing amino acid(s)being the most prominent.As a consequence,a thorough in-house quality characterization of peptides,already used in basic biomedical research,is mandatory. Remarkably,there is a large discrepancy between the certificates of analysis provided by the suppliers and the in-house quality control,which can at least partly be explained by the methods used.Finally,researchers should also be aware that the main peak observed in the chromatogram might be an impurity instead of the requested peptide;hence MS is requested in this QC.
Acknowledgments
This research was funded by PhD grants of"Institute for the Promotion of Innovation through Science and Technology in Flanders(IWT-Vlaanderen)"(Grant nos.131356 to F.V.and 101529 to M.D.)and The Special Research Fund(BOF)of Ghent University(Grant no.01J22510 to B.D.S.and E.W.).
[1]D.Craik,D.Fairlie,S.Liras,et al.,The future of peptide-based drugs, Chem.Biol.Drug Des.81(2013)136-147.
[2]R.Berggren,M.Moller,R.Moss,et al.,Outlook for the next 5 years in drug innovation,Nat.Rev.Drug Discov.11(2012)435-436.
[3]P.Van Arnum,Peptides gain traction in drug development,Pharm. Technol.36(2012)42-43.
[4]F.Albericio,H.Kruger,Therapeutic peptides,Future Med.Chem.4 (2012)1527-1531.
[5]W.Danho,J.Swistok,W.Khan,et al.,Peptides for Youth,in:S.Del Valle,E.Escher,W.Lubell(Eds.),Proceedings of the 20th American Peptide Symposium,SpringerSciencesand BusinessMedia, NewYork,NY,USA,2009,pp.467-468.
[6]P.Vlieghe,V.Lisowski,J.Martinez,et al.,Synthetic therapeutic peptides:science and market,Drug Discov.Today 15(2010) 40-56.
[7]P.Williams,Quorum sensing,communication and cross-kingdom signaling in the bacterial world,Microbiology 153(2007)3923-3938.
[8]D.T.Hughes,V.Sperandio,Inter-kingdom signaling:communication between bacteria and their hosts,Nat.Rev.Microbiol.6(2008)111-120.
[9]B.LaSarre,M.J.Federle,Exploiting quorum sensing to confuse bacterial pathogens,Microbiol.Mol.Biol.Rev.77(2013)73-111.
[10]E.Wynendaele,B.Gevaert,S.Stalmans,et al.,Exploring the chemical space of quorum sensing peptides(2015),Submitted for publication.
[11]T.Bruckdorfer,O.Marder,F.Albericio,From production of peptides in milligram amounts for research to multi-tons quantities for drugs of the future,Curr.Pharm.Biotechnol.5(2004)29-43.
[12]R.Merrifield,Solid phase peptide synthesis.I.The synthesis of a tetrapeptide,J.Am.Chem.Soc.85(1963)2149-2154.
[13]F.Guzman,S.Barberis,A.Illanes,Peptide synthesis:chemical or enzymatic,J.Biotechnol.10(2007)279-314.
[14]M.D’Hondt,N.Bracke,L.Tavernier,et al.,Related impurities in peptide medicines,J.Pharm.Biomed.Anal.101(2014)2-30.
[15]D.Takahashi,T.Yamamoto,Development of an efficient liquidphase peptide synthesis protocol using a novel fluorine-derived anchor support compound with fmoc chemistry;AJIPHASE®, Tetrahedron Lett.53(2012)1936-1939.
[16]M.Stawikowski,G.B.Fields,Introduction to peptide synthesis,Curr. Protoc.Protein Sci.18(2012)1-17.
[17]R.Dolling,M.Beyermann,J.Haenel,et al.,Innovation and Perspectives in Solid Phase Synthesis,in:R.Epton(Ed.),Proceedings of the 3rd International Symposium on Innovation and Perspectives in SPPS, Oxford UK,Mayflower Scientific Ltd.,Birmingham,1994,p.489.
[18]Y.Yang,W.V.Sweeney,K.Schneider,et al.,Aspartimide formation in base-driven 9-fluorenylmethoxycarbonyl chemistry,Tetrahedron Lett.35(1994)9689-9692.
[19]M.Hoitink,J.Beijnen,M.Boschma,et al.,Identification of the degradation products of gonadorelin and three analogues in aqueous solution,Anal.Chem.69(1997)4972-4978.
[20]V.Sanz-Nebot,F.Benavente,A.Castillo,et al.,Liquid chromatography-electrospray mass spectrometry of multicomponent peptide mixtures.Characterization of a mixture from the synthesis of the hormone goserelin,J.Chromatogr.A 889(2000)119-133.
[21]D.Riester,K.Wiesmüller,D.Stoll,et al.,Racemization of amino acids in solid-phase synthesis investigated by capillary electrophoresis,Anal.Chem.68(1996)2361-2365.
[22]B.De Spiegeleer,V.Vergote,A.Pezeski,et al.,Impurity profiling quality control testing of synthetic peptides using liquid chromatography-photodiode array-fluorescence and liquid chromatography-electrospray ionization-mass spectrometry:the obestatin case, Anal.Biochem.376(2008)229-234.
[23]V.Sanz-Nebot,B.Fernando,I.Toro,et al.,Separation and characterization of complex crude mixtures produced in the synthesis of therapeutic peptide hormones by liquid chromatography coupled to electrospray mass spectrometry(LC-ESI-MS),Anal.Chim.Acta 521 (2004)25-36.
[24]I.Eggen,F.Bakelaar,A.Petersen,et al.,A novel method for repetitive peptide synthesis in solution without isolation of intermediates,J.Peptide Sci.11(2005)633-641.
[25]H.Ball,P.Mascagni,Chemical synthesis and purification of proteins: a methodology,Int.J.Pept.Protein Res.48(1996)31-47.
[26]J.W.Metzger,C.Kempter,K.H.Wiemuller,et al.,Electrospray mass spectrometry and tandem mass spectrometry of synthetic multicomponent peptide mixtures:determination of composition and purity,Anal.Biochem.219(1994)261-277.
[27]D.S.King,C.G.Fields,G.B.Fields,A cleavage method which minimizes side reactions following fmoc solid phase peptide synthesis,Int.J.Pept.Protein Res.36(1990)255-266.
[28]F.Albericio,N.Kneib-Cordonier,S.Biancalana,et al.,Preparation and application of the 5-(4-(9-fluorenylmethyloxycarbonyl)aminomethyl-3,5-dimethoxyphenoxy)valeric acid(PAL)handle for the solid-phase synthesis of C-terminal peptide amides under mild conditions,J.Org.Chem.155(1990)3730-3743.
[29]N.A.Solé,G.Barany,Optimization of solid-phase synthesis of [Ala8]-dynorphin A,J.Org.Chem.57(1992)5399-5403.
[30]J.Leon,E.Reubsaet,H.Beijnen,et al.,Reduction of Cys36-Cys42 and Cys64-Cys74disulfide bonds in recombinant human granulocyte colony stimulating factor,J.Pharm.Biomed.Anal.19(1999)837-845.
[31]M.Verbeken,E.Wynendaele,R.Lefebre,et al.,The influence of peptide impurity profiles on functional tissue-organ bath response:the 11-mer peptide INSL6[151-161]case,Anal.Biochem.421(2012) 547-555.
[32]WHO,Handbook:Quality Practices in Basic Biomedical Research,〈http:// www.who.int/tdr/publications/documents/quality_practices.pdf〉,2006.
[33]J.de Beukelaar,J.Gratama,P.Smitt,et al.,The impact of impurities in synthetic peptides on the outcome of T-cell stimulation assays, Rapid Commun.Mass Spectrom.21(2007)1282-1288.
[34]J.Currier,L.Galley,H.Wenschuh,et al.,Peptide impurities in commercial synthetic peptides and their implications for vaccine trial assessment,Clin.Vaccine.Immunol.15(2008)267-276.
[35]J.Zhang,C.Klein,P.Ren,et al.,Response to comment on obestatin, a peptide encoded by the ghrelin gene,opposes ghrelin’s effect on food intake,Science 315(2007)766d.
[36]E.Wynendaele,F.Verbeke,M.D’Hondt,Crosstalk between the microbiome and cancer cells by quorum sensing peptides,Peptides 64 (2015)40-48.
[37]B.De Spiegeleer,F.Verbeke,M.D’Hondt,et al.,The quorum sensing peptides PhrG,CSP and EDF promote angiogenesis and invasion of breast cancer cells in vitro,PLoS One(2015)http://dx. doi.org/10.1371/journal.pone.0119471.
[38]E.Wynendaele,A.Bronselaer,J.Nielandt,et al.,Quorumpeps database:chemical space,microbial origin and functionality of quorum sensing peptides,Nucleic Acids Res.41(2013)D655-D659. [39]European Pharmacopoeia 7.0,European Directorate for the quality of Medicinesand Healthcare,Strassbourg,France,07/2009:2034, Substances for Pharmaceutical Use.
[40]OMCL Network of the Council of Europe,Qualification of equipment,Annex 1:Qualification of HPLC Equipment,PA/PH/OMCL (11)04,2011.
[41]P.R.Baker,K.R.Clauser〈http://prospector.ucsf.edu.〉,acccessed 24/09/2014.
[42]P.Roepstorff,J.Fohlman,Proposal for a common nomenclature for sequence ions in mass spectra of peptides,Biomed.Mass Spectrom. 11(1984)601.
[43]M.C.Lai,E.M.Topp,Solid-state chemical stability of proteins and peptides,J.Pharm.Sci.88(1999)489-500.
[44]V.Sanz-Nebot,A.Garcés,J.Barbosa,Investigation of crudes of synthesis of carbetocin by liquid chromatography coupled to electrospray ionization mass spectrometry,J.Chromatogr.A 883(1999) 267-275.
[45]A.Taicrib,G.K.Scriba,C Neusüß,Identification and characterization of impurities of tetracosactide by capillary electrophoresis and liquid chromatography coupled to time-of-flight mass spectrometry,Anal. Bioanal.Chem.401(2011)1365-1375.
✽Corresponding author.Tel.:+32 9 264 81 00;fax:+32 9 264 81 93. E-mail address:Bart.DeSpiegeleer@UGent.be(B.De Spiegeleer).1These authors contributed equally to this work.
Peer review under responsibility of Xi’an Jiaotong University.
2095-1779©2015 Xi’an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BYNC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Impurity profiling
 Journal of Pharmaceutical Analysis2015年3期
Journal of Pharmaceutical Analysis2015年3期
- Journal of Pharmaceutical Analysis的其它文章
- Non-covalent binding analysis of sulfamethoxazole to human serum albumin:Fluorescence spectroscopy,UV-vis,FT-IR,voltammetric and molecular modeling
- Determination of diclofenac in pharmaceutical preparations by voltammetry and gas chromatography methods
- Comparison of conventional and supported liquid extraction methods for the determination of sitagliptin and simvastatin in rat plasma by LC-ESI-MS/MS
- Extraction,characterization and biological studies of phytochemicals from Mammea suriga
- Liquid chromatography-tandem mass spectrometry method for the estimation of adefovir in human plasma:Application to a pharmacokinetic study
- Selective extraction of dimethoate from cucumber samples by use of molecularly imprinted microspheres