
动物源食品中β受体阻断剂残留量测定的不确定度评定
2015-12-07 12:35张鸿伟王凤美
质谱学报 2015年2期
许 辉,张鸿伟,张 罡,王凤美
(山东出入境检验检疫局,山东青岛 266002)
动物源食品中β受体阻断剂残留量测定的不确定度评定
许 辉,张鸿伟,张 罡,王凤美
(山东出入境检验检疫局,山东青岛 266002)
建立了液相色谱-串联质谱(LC-MS/MS)法测定动物源食品中β受体阻断剂残留量的数学模型,依据影响测量结果的不确定度来源,如标准溶液配制、样品称量、回归方程、方法重复性等,对各个不确定度分量进行评定和分析,给出了0.5μg/kg检测水平下,液相色谱-串联质谱法测定食品中β受体阻断剂残留量的相对合成标准不确定度以及扩展不确定度。通过比较各分量不确定度,发现实验过程中最小二乘法拟合带来的不确定度影响最大。
不确定度;β受体阻断剂;液相色谱-串联质谱(LC-MS/MS)
不确定度是表征合理地赋予被测量值的分散性,与测量结果相联系的参数[1]。一个完整的测量结果,除了给出被测量的最佳估计值之外,还应同时给出测量结果的不确定度。建立和实施各类检测的不确定度评定方法,既是提高检测质量的要求,也是实现检测数据国际互认所不可缺少的内容[2-9]。
β受体阻断剂是能选择性地与β肾上腺素受体结合,从而拮抗神经递质和儿茶酚胺对β受体激动作用的一种药物类型。受体阻断剂具有负性变时、变力作用,可引起心动过缓、房室传导阻滞,诱发心力衰竭或心功能恶化,使白细胞减少,血尿酸升高[10-16]。目前,液相色谱-串联质谱法(LC-MS/MS)测定动物源食品中β受体阻断剂残留量的不确定度评定尚未见报道。
本工作按照JJ1059-2012规定,对液相色谱-串联质谱法测定动物源食品中β受体阻断剂残留量的不确定度来源进行分析。根据所建立的数学模型,对各个分量的不确定度进行评估,得到本实验的不确定度,以便为科学地评定测量结果提供依据。
1 实验部分
1.1 主要仪器与试剂
Agilent 1290液相色谱系统:美国Agilent公司产品;API 5500三重四极杆质谱分析仪:美国AB Sciex公司产品;T25basic高速均质器、涡旋混匀器、HS260basic水平振荡器:德国IKA公司产品;CR22GⅡ高速冷冻离心机:日本日立公司产品;Turbo Vap LV型氮吹浓缩仪:美国Caliper公司产品。
阿替洛尔、美托洛尔、卡拉洛尔、卡拉洛尔-D7、比索洛尔:均为德国Dr.Ehrenstorfer公司产品;阿替洛尔-D7、吲哚洛尔、吲哚洛尔-D7、美托洛尔-D7、醋丁洛尔-D5、比索洛尔-D5、拉贝洛尔:均为加拿大TRC公司产品;普萘洛尔:美国Sigma公司产品;普萘洛尔-D9:加拿大CDN公司产品;喷布洛尔:EDQM公司产品;喷布洛尔-D9:德国Witega公司产品;醋丁洛尔:美国Fluka公司产品;甲酸(色谱级):美国Riedel-de Haen公司产品;甲醇、乙腈、正己烷(色谱级):B&J Brand公司产品;实验用水:由Millipore系统制得的超纯水。
1.2 样品前处理
称取(2.00±0.01)g粉碎均匀的试样,置于50mL离心管中,分别加入40μL 0.1mg/L混合内标溶液、40μL酶解液和5mL EDTA提取液,涡旋混匀1min,高速均质器均质,置于50℃水浴振荡器中恒温振荡60min,调节pH值。分别加入2g硅藻土、2g NaCl和25mL乙腈,振荡10min,以10 000r/min离心10min,移取上清液,过净化填料,涡旋混匀1min,振荡5min,以10 000r/min离心5min,移取5mL上清液于40℃下氮气吹干。用1mL甲醇-0.2%甲酸水(1∶9,V∶V)复溶,过0.22μm滤膜,供液相色谱-串联质谱仪测定。
2 不确定度评定
2.1 数学模型
根据内标法,测量计算公式如下:
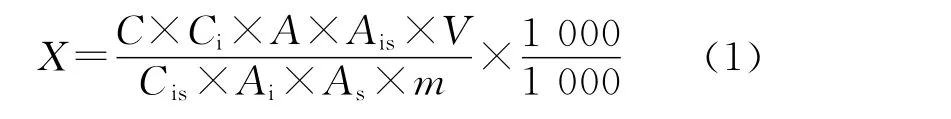
式中:X为试样中待测组分的残留量(μg/kg);C为标准工作溶液的浓度(μg/L);Cis为标准工作溶液中内标物的浓度(μg/L);Ci为样液中内标物的浓度(μg/L);A为样液中待测目标物的峰面积;As为标准工作溶液中目标物的峰面积;Ais为标准工作溶液中内标物的峰面积;Ai为样液中内标物的峰面积;V为样品定容体积(mL);m为样品称样量(g)。
实验中,标准工作溶液中内标物浓度Cis和样液中内标物浓度Ci带来的不确定度,以及峰面积A、As、Ais、Ai的不确定度,可以并入到由标准曲线求得的样液中待测物与其同位素内标物的质量浓度比x0中;frep为测量重复性影响因素的修正因子,其值为1,因此式(1)可改为:
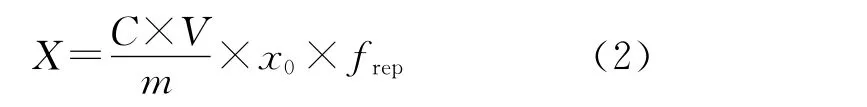
2.2 不确定度来源
从测量过程和数学模型分析,样品测定的不确定度来源主要有:标准溶液配制引入的不确定度urel(C);标准曲线校准引入的不确定度urel(x0);样品称量引入的不确定度urel(m);样品溶液添加、稀释和定容引入的不确定度urel(V);结果重复性引入的不确定度urel(frep)。不确定度来源示意图示于图1。
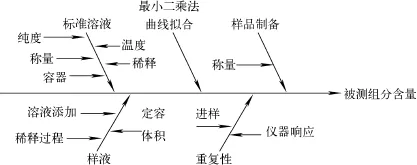
图1 不确定度来源示意图Fig.1 The schematic of uncertainty source
上述各参数影响相互独立,因此其相对合成标准不确定度urel(X)可表征为以上5项不确定度的平方和再开二次方。
2.3 各分量不确定度的计算
2.3.1 标准溶液配制引入的不确定度urel(C)
准确称取各约10mg 9种标准品,分别移入10mL容量瓶中,用甲醇稀释至刻度,配成1 000mg/L标准储备溶液;从中分别移取100μL于10mL容量瓶中,用甲醇定容,得到浓度为10mg/L混合标准中间液;从混合标准中间液中移取100μL于10mL容量瓶中,用甲醇定容,得到浓度为100μg/L混合标准工作液。按如上操作,称取一定量的内标标准品,分别获得浓度为1 000mg/L、10mg/L、100μg/L内标混合溶液。
标准溶液的配制包括β受体阻断剂类标准溶液的配制和β受体阻断剂类内标溶液的配制,因此标准溶液的相对标准不确定度为:urel(C)=以吲哚洛尔为例,其标准溶液引入的不确定度评定如下:
1)标准溶液配制引入的不确定度
(1)吲哚洛尔纯度为98.0%,其相对不确
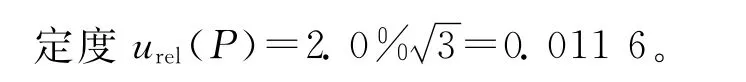
(2)标准品称量使用万分之一天平,精确至0.1mg。天平校准证书给出的不确定度为0.17mg,k=2,则校准产生的标准不确定度为u1(w)=0.17/2=0.985mg;利用《化学分析中不确定度的评估指南》[7]附录G中给定的数据,对十万分之一分析天平最后一位有效数字为0.01mg,则由天平重复性产生的不确定度为u2(w)=0.5×0.01=0.05mg。标准品称样量为10.2mg,因此,标准品称量带来的相对标准不确定度为0.008 35。
(3)标准品配制所使用的10mL A级容量瓶20℃时的容量允差为±0.020mL,服从三角分布,区间半宽度此引起的标准不确定度u1(V10)=a(V10)/k=0.008 17mL;实验室温度在(20±5)℃之间变动,甲醇的膨胀系数(20℃)为0.001 18/℃,对10mL容量瓶由温度效应产生的体积变化ΔV=10×5×0.001 18=0.059mL,服从均匀分布,由此引起的标准不确定度u2(V10)=a/k=0.034 1mL,故:
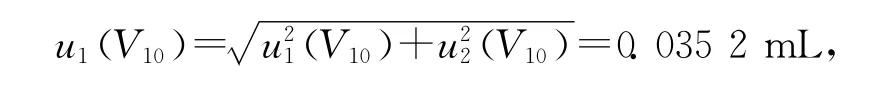
其相对标准不确定度urel(V1)=u(V10)/V10=0.003 52。
使用100μL移液枪稀释标准溶液,其容量允差为±1.0μL,服从均匀分布,由此引入的标准不确定度为u1(V100)=a1(V100)/k=0.057 8μL;由温度效应产生的体积变化ΔV=100×5×0.001 18=0.590μL,服从均匀分布,,故稀释过程中使用移液枪引入的标准不确定度为稀释过程中,使用10mL容量瓶和移液枪各2次,其中,10mL容量瓶的合成标准不确定度为u(V10)=0.035 2mL,移液枪移取的体积均为100μL,故标准溶液稀释过程引入的相对标准不确定度为:
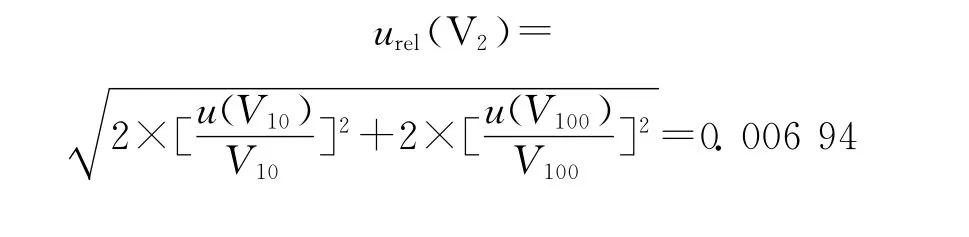
由此,标准溶液配制、稀释过程引入的相对标准不确定度为:
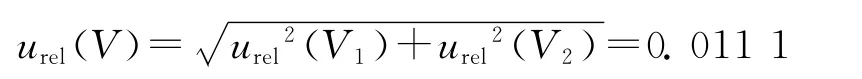
综上,吲哚洛尔标准溶液配制引入的相对标准不确定度为:
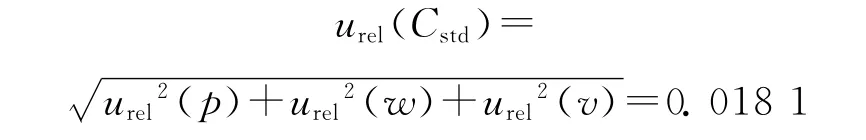
2)内标溶液配制引入的不确定度
(2)吲哚洛尔-D7称量引入的不确定度计算过程与标准溶液引入的不确定度相同,称样量为10.3mg,故其相对合成标准不确定度urel(w)=
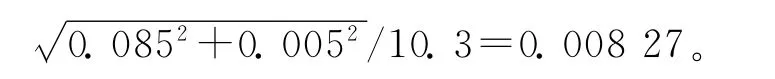
(3)吲哚洛尔-D7溶液配制和稀释过程中,相对不确定度计算与标准溶液的计算方法相同,其值分别为urel2(V1)=0.003 52、urel2(V2)=0.006 94;实验过程中,使用100μL移液枪添加内标溶液,移液枪引入的标准不确定度为u(V100)=0.671μL,样品前处理过程中内标溶液的移取体积为40μL,故内标溶液添加引入的相对标准不确定度为urel(V3)=0.671/40= 0.016 8。
由此,内标配制、稀释、添加过程引入的相对标准不确定度为:
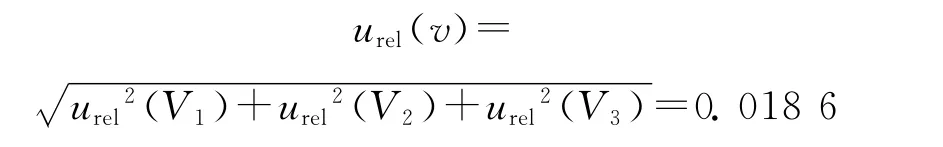
故吲哚洛尔-D7内标溶液配制引入的相对标准不确定度为:
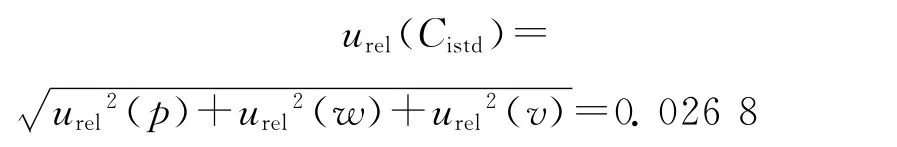
标准溶液配制引入的相对标准不确定度为:
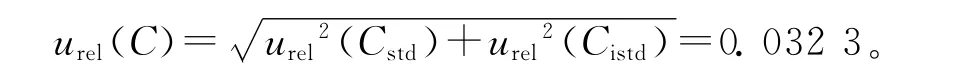
9种β受体阻断剂类药物标准溶液引入的相对不确定度列于表1。
2.3.20.2、0.5、0.8μg/L,其内标浓度均为0.08μg/L。对β受体阻断剂类药物与其内标质量浓度比x0分别为0、0.125、0.25、0.5、1.0、2.0、5.0、10.0的8个校准溶液各平行测定3次,得到相应的标准物质与内标物质峰面积比值y;对以上测定数据采用最小二乘法进行拟合,得到校准曲线方程y=bx+a和拟合系数r2,相关结果列于表2。
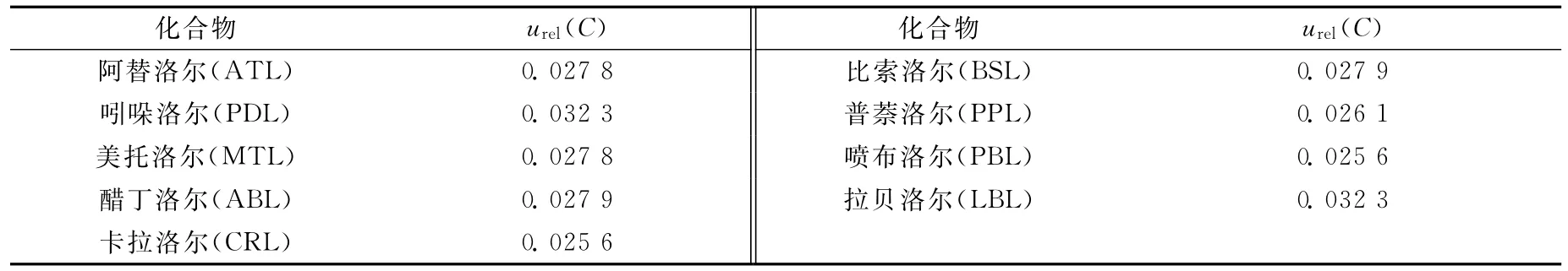
表1 标准溶液引入的相对不确定度Table 1 The relative uncertainty of standard solutions
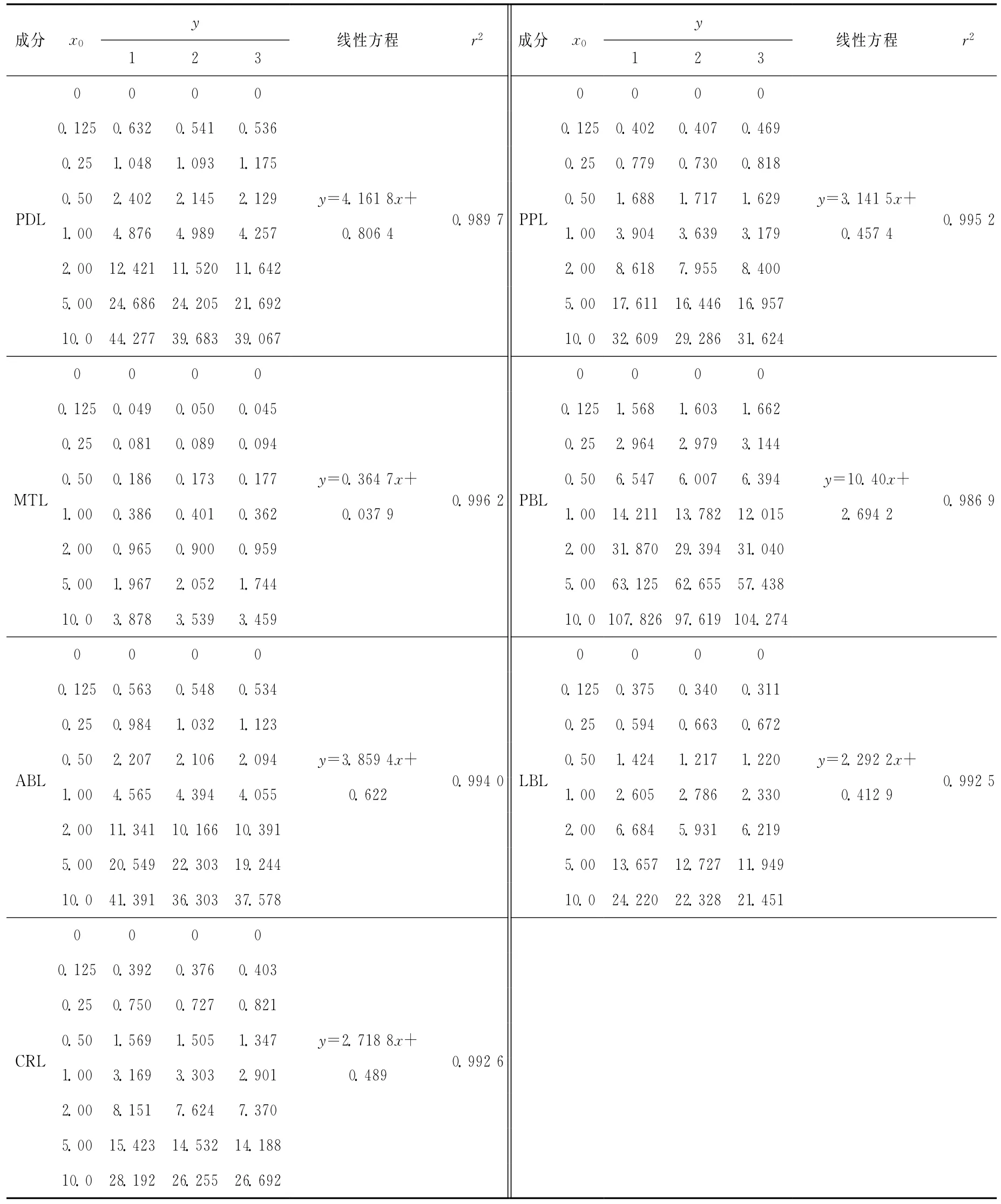
续表2
为了评定最小二乘法估计量的不确定度,首先必须对直接测量所得的测量数据进行不确定度估计,实际上是求出测量数据的实验标准差:
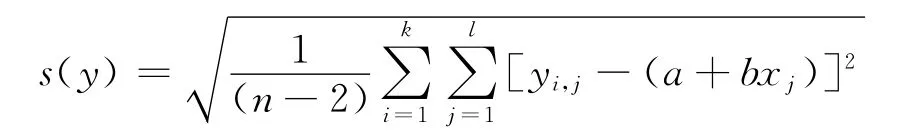
式中,[yi,j-(a+bxj)]为当输入量为xj时,仪器响应值与拟合直线上对应的响应值之差。
使用间接式测量仪器测量时,利用校准曲线,在测得样品响应值y0后,可按线性回归方程计算被测量的预估值x0,其标准不确定度为:
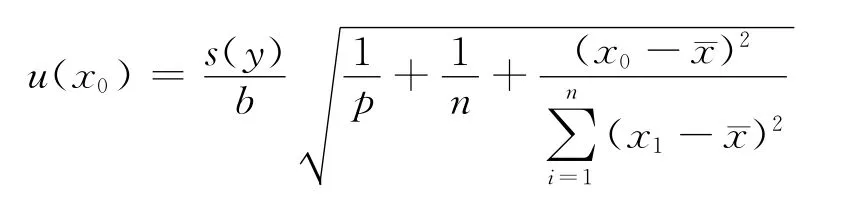
式中,s(y)为校准溶液峰面积比残差的标
准偏差;b为拟合直线的斜率;p为样品平行测量次数,本例为6;n为拟合直线的数据对总数,本例为24;x0为样品平行测量P次结果的平均值;¯x为绘制拟合直线全部(n个)输入值x1的总平均值,urel(x0)=u(x0)/x0,计算结果列于表3。 2.3.3 样品称量引入的不确定度 使用百分之一天平称量样品,称样量为2.00g,精确至0.01g。天平校准证书给出的不确定度为0.005 80g,k=2,故u(m)=0.005 80/2=0.002 90g,其相对标准不确定度为urel(m)=u(m)/m=0.001 45。
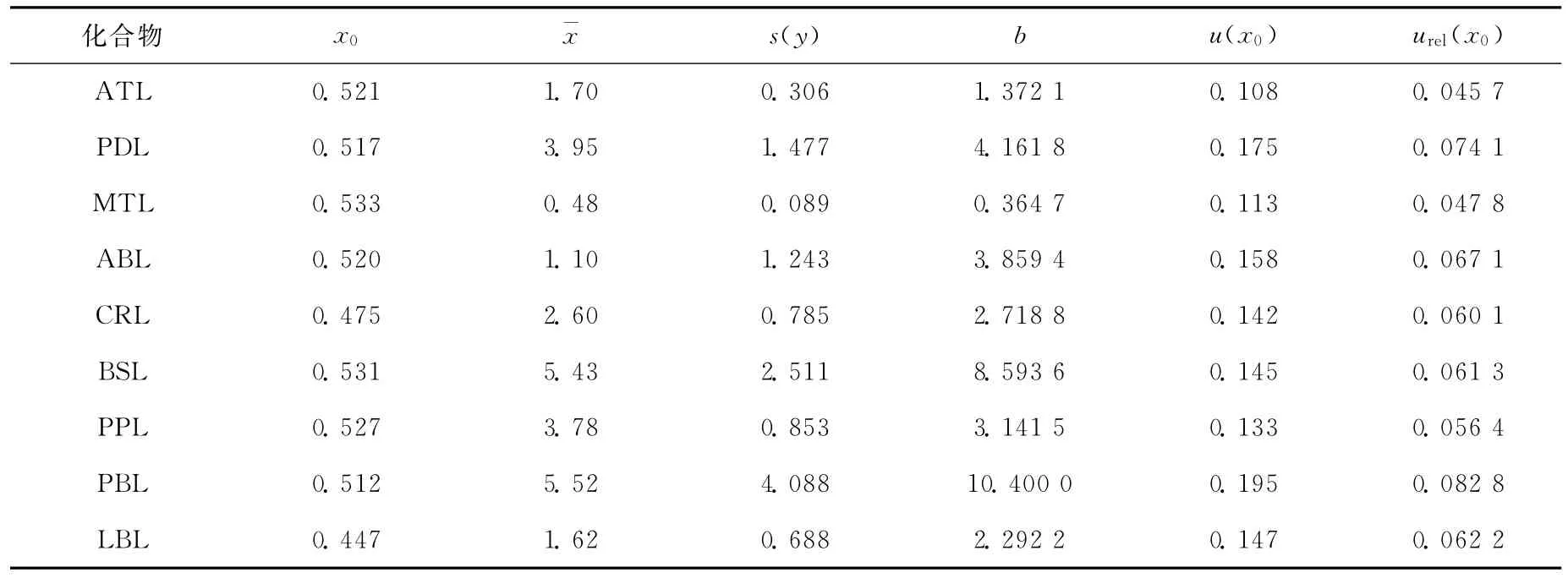
表3 标准曲线引入的不确定度Table 3 The uncertainty of standard curve
2.3.4样品溶液添加、稀释和样品定容引入的不确定度 实验过程中,使用25.0mL可调加液器添加乙腈提取液。容量允差为±100.0 μL,服从均匀分布mL;由温度效应产生的体积变化为ΔV= 0.171 2mL,服从均匀分布,其相对标准不确定度urel(V1)=u(V10)/V10=0.011 5。
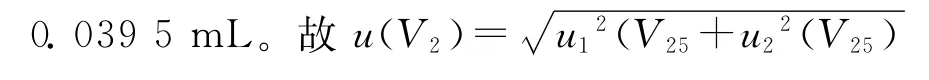
实验过程中,使用5.0mL移液枪移取氮吹浓缩的5mL样品溶液。移液枪的容量允差为±50.0μL,服从均匀分布28.9μL;由温度效应产生的体积变化为ΔV= 34.25μL,服从均匀分布,其相对标准不确定度为urel(V2)=u(V5000)/V5000=0.011 5。
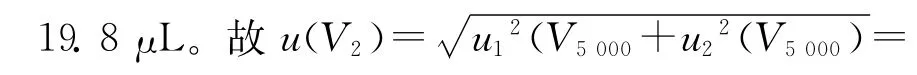
样品定容使用1 000μL移液枪,其容量允差为±10.0μL,服从均匀分布,u1(V1000)=a(V1000)/k=5.78μL;实验室温度引起的体积变化可以忽略不计,故urel(V3)=u(V1000)/V1000=0.005 78。
综上,样品溶液添加、稀释和样品定容引入的不确定度为:
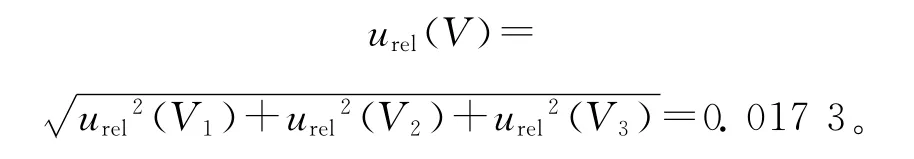
2.3.5 结果重复性引入的不确定度 在重复性条件下,对样品进行了n次独立测试(n=6)。以吲哚洛尔为例,平行测定结果分别为:0.494、0.574、0.581、0.465、0.508、0.477μg/kg,平均值为根据贝塞尔公式计算得单次测量的标准不确定度u(X)=
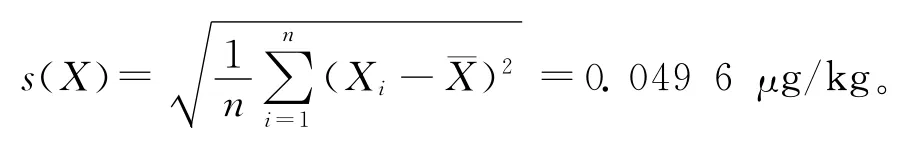
以多次测量结果的算术平均值为最佳估算值,则测定结果的标准不确定度u(¯X)=s(¯X)=由此引起的结果重复性引入的相对标准不确定度0.039 1。由结果重复性引入的9种β受体阻断剂类药物的相对标准不确定度列于表4。

表4 结果重复性引入的相对标准不确定度Table 4 The relative uncertainty of repeatability of results
3 样品合成标准不确定度、扩展不确定度
由2.2节求得各物质的相对合成不确定度,则合成标准不确定度u(X)=X×urel(X);取包含因子k=2,置信概率p=95%,求得扩展不确定度为U(X)=k×u(X),具体结果列于表5。
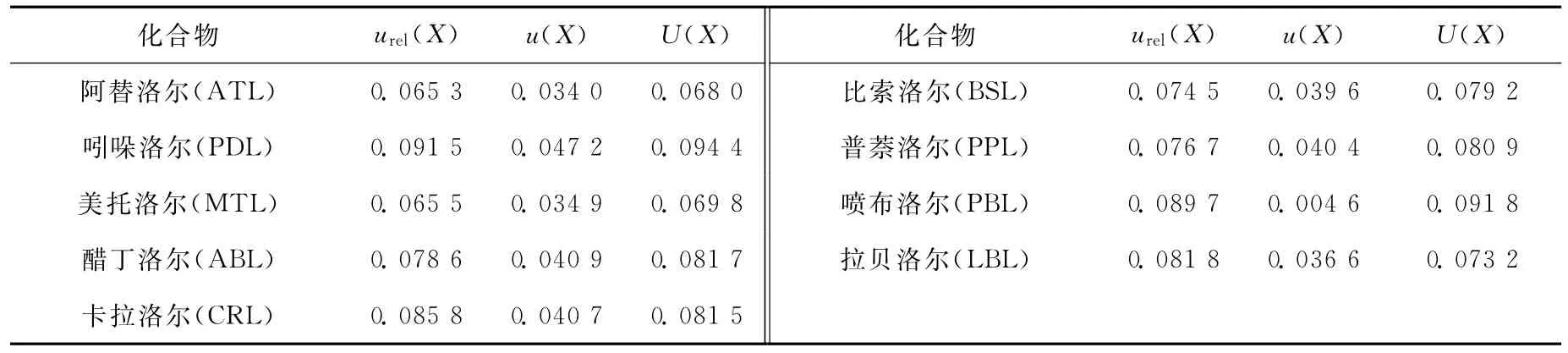
表5 9种β受体阻断剂类药物的不确定度Table 5 The uncertainty of 9 kinds of beta-blockers
4 结论
本工作采用液相色谱-串联质谱法对食品中9种β受体阻断剂的残留量进行测定,比较各分量不确定度大小,发现实验过程中最小二乘法拟合带来的不确定度影响最大;标准溶液的配制、进样过程、仪器响应和测量的重复性对不确定度有一定的贡献,而样品称量的不确定度可以忽略不计。
[1] JJF 1059.1-2012.测量不确定度评定与表示[S].北京:中国标准出版社,2012.
[2] CNAS-GL06.化学分析中不确定度的评价指南[S].北京:中国标准出版社,2006.
[3] 朱绍棠,徐友宣,吴侔天,等.用HPLC法测定咖啡因的不确定度[J].分析测试学报,2003,22(5):88-90.ZHU Shaotang,XU Youxuan,WU Moutian,et al.Uncertainty measurement in the HPLC determination o f caffeine[J].Journal of Instrumental Analysis,2003,22(5):88-90(in Chi-nese).
[4] 刘海山,陈笑梅,李蕾蕾,等.液相色谱串联质谱法测定水产品中硝基呋喃类代谢物的不确定度评定[J].分析试验室,2009,28(S2):187-191.LIU Haishan,CHEN Xiaomei,LI Leilei,et al.Evaluating the uncertainty of nitrofuran antibiotics and metabolites in aquatic product using LCMS/MS[J].Chinese Journal of Analysis Laboratory,2009,28(S2):187-191(in Chinese).
[5] ELLISON S L R,ROSSLEIN M,WILLIAMS A.Quantifying uncertainty in analytical measurement,2th Edition[M].EURACHEM/CITAC,2000:1-257.
[6] LEITO S,MÖLDER K,KÜNNAPAS A,et al.Uncertainty in liquid chromatographic analysis of pharmaceutical product:Influence of various uncertainty sources[J].Journal of Chromatography A,2006,1 121(1):55-63.
[7] JJF 1135-2005.化学分析测量不确定度评定[S].北京:中国标准出版社,2005.
[8] 陈晓翔,邹永德,王长安,等.液相色谱串联质谱法测定动物源食品中克伦特罗残留量的不确定
度评定[J].计量学报,2012,33(4):372-376.CHEN Xiaoxiang,ZOU Yongde,WANG Changan,et al.Uncertainty evaluation for determination of clenbuterol residues in foodstuffs of animal origin by liquid chromatography tandem mass spectrometry[J].Acta Metrologica Sinica,2012,33(4):372-376(in Chinese).
[9] 严 凤,张文刚,李丹妮,等.液相色谱-串联质谱法测定动物源食品中氯霉素残留量不确定度分析[J].上海畜牧兽医通讯,2008,(5):32-33.YAN Feng,ZHANG Wengang,LI Danni,et al.Uncertainty evaluation for determination of residual quantity of chloramphenicol in foodstuffs of animal origin by liquid chromatography tandem mass spectrometry[J].Shanghai Journal of Animal Husbandry and Veterinary Medicine,2008,(5):32-33(in Chinese).
[10]MCMURRAY J J V,KENDALL M J.Betablockers in heart failure[M].London:Martin Dunitz Ltd,2002.
[11]SILVA L C,TREVISAN M G,POPPI R J,et al.Direct determination of propranolol in urine by spectrofluorimetry with the aid of second order advantage[J].Anal Chim Acta,2007,595(1/2):282-288.
[12]MITROWSKA K,POSYNIAK A,ZMUDZKI J.Multiresidue method for the determination of nitroimidazoles and their hydroxy-metabolites in poultry muscle,plasma and egg by isotope dilution liquid chromatography-mass spectrometry[J].Anal Chim Acta,2009,637(1/2):185-188.
[13]ANASTASSIADES M,LEHOTAY S J,STA-JNBAHER D,et al.Fast and easy multiresidue method employing acetonitrile extraction/partitioning and“dispersive solid-phase extraction”for the determination of pesticide residues in produce[J].J AOAC Int,2003,86(2):412-431.
[14]张鸿伟,简慧敏,林黎明,等.液相色谱-四极杆/离子阱质谱快速测定蜂蜜中痕量硝基咪唑类药物及其代谢物残留[J].分析测试学报,2012,31(7):763-770.ZHANG Hongwei,JIAN Huimin,LIN Liming,et al.Rapid detection of trace amounts of nitroimidazoles and their metabolites in honey using liquid chromatography coupled with quadrupole/linear ion trap mass spectrometry[J].Journal of Instrumental Analysis,2012,31(7):763-770(in Chinese).
[15]张鸿伟,蔡雪,林黎明,等.液相色谱-四极杆/离子阱质谱同时确证和测定肌肉中16种同化甾体激素残留[J].色谱,2012,30(10):991-1 001.ZHANG Hongwei,CAI Xue,LIN Liming,et al.Simultaneous identification and detection of 16 anabolic steroid hormones in muscle using liquid chromatography coupled to quadrupole/linear ion trap mass spectrometry[J].Chinese Journal of Chromatography,2012,30(10):991-1 001(in Chinese).
[16]O’MAHONY J,CLARKE L,WHELAN M,et al.The use of ultra-high pressure liquid chromatography with tandem mass spectrometric detection of analysis of agrochemical residues and mycotoxines in food-challenges and applications[J].J Chromatogr A,2013,1 292:83-89.
Uncertainty Evaluation in the Determination of β-Blockers in Food of Animal Origin
XU Hui,ZHANG Hong-wei,ZHANG Gang,WANG Feng-mei
(Shandong Entry-Exit Inspection Quarantine of the People’s Republic of China,Qingdao 266002,China)
β-Blockers in food of animal origin was determined by liquid chromatographytandem mass spectrometry(LC-MS/MS).According to the mathematic model,the uncertainty sources of test results were analyzed,such as the standard solution preparation,sample weighing,regression equation,method repeatability.Each component uncertainty was calculated through analyzing.With the limits of quantification of 0.5μg/kg for all analytes,the expanded uncertainty was calculated while the combined standard uncertainty ofβ-blockers residue was obtained.Comparing of each component of uncertainty in the process,the results show that the effect of the least squares fitting is the maximun.
uncertainty;β-blockers;liquid chromatography-tandem mass spectrometry(LC-MS/MS)
O657.63
A
1004-2997(2015)02-0185-08
10.7538/zpxb.youxian.2014.0062
2014-03-16;
2014-07-07
国家质检总局科技项目(2007IK144);山东省科技发展计划项目(2008GG10009020);山东出入境检验检疫局科研攻关项目(SK200820)资助
许 辉(1982—),男(汉族),工程师,从事药物残留检验研究。E-mail:xh2k12@sohu.com
时间:2014-12-02;
http:∥www.cnki.net/kcms/doi/10.7538/zpxb.youxian.2014.0062.html
猜你喜欢
浙江化工(2022年1期)2022-02-19
口腔护理用品工业(2021年4期)2021-11-02
化工设计通讯(2020年10期)2020-09-17
中成药(2018年6期)2018-07-11
科教导刊(2017年26期)2017-11-07
科技与创新(2015年17期)2015-09-11
天津体育学院学报(2015年6期)2015-06-01
科技与创新(2014年12期)2014-08-28
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
中国信息化·学术版(2013年3期)2013-06-25
