
N,N′-二-[3-羟基-4-(2-苯并噻唑)苯基]脲的光谱实验与密度泛函理论研究
2015-12-05 06:29崔颖娜尹静梅贾颖萍李慎敏大连大学环境与化学工程学院辽宁大连66大连大学生物有机省重点实验室辽宁大连66
物理化学学报 2015年9期
崔颖娜 尹静梅 贾颖萍 李慎敏,*(大连大学环境与化学工程学院, 辽宁 大连 66; 大连大学生物有机省重点实验室, 辽宁 大连 66)
N,N′-二-[3-羟基-4-(2-苯并噻唑)苯基]脲的光谱实验与密度泛函理论研究
崔颖娜1,*尹静梅1,2贾颖萍1李慎敏1,2,*
(1大连大学环境与化学工程学院, 辽宁 大连 116622;2大连大学生物有机省重点实验室, 辽宁 大连 116622)
利用实验观测与密度泛函理论(DFT)计算方法考察了新化合物N,N′-二-[3-羟基-4-(2-苯并噻唑)苯基]脲(4-DHBTU)的红外、核磁与紫外吸收光谱性质. 与单体2-(4-氨基-2-羟苯基)苯并噻唑(4-AHBT)相比, 4-DHBTU的实验紫外吸收强度显著增强, 最大吸收峰发生了明显红移, 并呈现出双吸收峰特征. 结合实验光谱数据与密度泛函理论计算分析表明, 4-DHBTU分子最稳定的基态异构体为cis-C11和trans-C11, 而导致上述紫外光谱差异的主要原因是4-DHBTU样品中cis-C11, trans-C11, cis-C22, trans-C22等多种异构体共存. 此外, 4-DHBTU与溶剂二甲基亚砜(DMSO)间氢键作用使得核磁实验中4-DHBTU的15H、16H氢谱化学位移显著增大.
N,N′-二-[3-羟基-4-(2-苯并噻唑)苯基]脲; 紫外光谱; 激发态; 密度泛函理论
1 引 言
激发态分子内质子转移(ESIPT)化合物具有光学双稳态、非线性光学效应和光致变色等特性,1–3通常在传感器、荧光离子探针等领域有着重要的应用.4–7近年来,基于2-(2-羟苯基)苯并唑类的ESIPT 化合物的研究引起了人们的极大兴趣.8–172-(4-氨基-2-羟苯基)苯并噻唑(4-AHBT)是典型的ESIPT化合物, 然而, 由于4-AHBT的紫外吸收与荧光发射强度相对较弱, 在一定程度上限制了其应用.18为了得到具有更好光学性能的ESIPT化合物,最近, 我们利用N,N′-羰基二咪唑(CDI)易于脱去羰基的化学特性,19将其脱去的羰基与两分子的4-AHBT进行反应, 成功合成了新化合物N,N′-二-[3-羟基-4-(2-苯并噻唑)苯基]脲(4-DHBTU) (合成路线见图1).20作为新合成化合物, 目前针对4-DHBTU的研究几乎是空白. 简单的电子结构理论分析预示, 一方面, 脲官能团的引入将使4-DHBTU分子中π电子的共轭区域较单体4-AHBT成倍扩大, 进而增加分子的稳定性, 并导致其吸收光谱的红移; 另一方面,由于4-DHBTU分子具有两个4-AHBT, 即具有两个可以发生ESIPT过程的结构单元, 将有利于4-DHBTU分子光谱强度的增强.
本文中, 我们利用紫外光谱实验考察了乙腈溶液中4-AHBT与4-DHBTU的紫外吸收光谱. 光谱分析显示, 4-DHBTU的紫外吸收比4-AHBT有了很大的改善, 紫外吸收强度显著增强, 最大吸收峰发生了明显红移, 而且4-DHBTU的紫外吸收谱图中出现了双强吸收峰. 显然, 4-DHBTU独特的光谱性质与其结构有关. 由4-DHBTU的合成路线可知, 合成产物4-DHBTU也是光谱测量的样本, 很可能由几种4-DHBTU顺反异构体共同组成, 然而, 由于其难于分离, 仅用实验光谱解析来进行结构预测难度较大.
为了合理解析上述紫外光谱实验, 利用密度泛函理论方法, 对4-DHBTU分子可能的异构体进行了几何结构优化, 分别计算了红外(IR)光谱和核磁共振(NMR)氢谱, 通过与实验观测值的比对, 确定了4-DHBTU分子的稳定几何结构; 然后, 利用含时密度泛函理论(TD-DFT)方法,21,22计算了4-DHBTU分子的垂直跃迁能, 并与紫外吸收光谱实验进行了比较与讨论, 提出了实验条件下4-DHBTU样品为多种异构体共存, 进而揭示了4-DHBTU紫外吸收双峰的发生机制.

图1 4-DHBTU 的合成路线Fig.1 Synthetic routes for 4-DHBTU
2 实验部分
2.1 试剂与仪器
4-氨基水杨酸(98%), 邻苯二胺(98%), 多聚磷酸PPA (98%), N,N′-羰基二咪唑(98%), 上海晶纯实业有限公司; 所用溶剂均为分析纯, 使用前均经除水精制.
美国Nicolet-550型红外光谱仪, KBr压片; 德国Bruker公司AV500型核磁共振仪, DMSO-d6(以下简写为DMSO)为溶剂, 四甲基硅烷(TMS)为内标; 日本Jasco公司的V-560型UV-Vis分光光度计, 乙腈为溶剂.
2.2 实验过程
2.2.1 4-DHBTU的制备及红外、核磁表征
4-DHBTU的制备方法按文献20进行.
IR (KBr), ν/cm–1: 1217.56, 1397.03, 1436.99, 1474.22, 1545.06, 1597.50, 3060.44, 3275.65.
1H NMR (DMSO-d6, 500 MHz ) δ: 7.03 (2H, dd, J = 8.7, 2.0 Hz, ArH), 7.41 (2H, m, ArH), 7.45 (2H, d, J = 2.0 Hz, ArH), 7.52 (2H, m, ArH), 8.01 (2H, d, J = 8.0 Hz, ArH), 8.08 (2H, d, J = 8.7 Hz, ArH), 8.11 (2H, d, J = 7.9 Hz, ArH), 9.11 (2H, s, NH), 11.70 (2H, s, ArOH).
2.2.2 4-DHBTU的紫外光谱实验
分别配制浓度为5 × 10–2molL–1的4-DHBTU和4-AHBT乙腈溶液, 各自稀释至5×10–6molL–1, 以乙腈溶剂为空白, 测定它们在280–400 nm区间的紫外光谱. 将溶液放置24 h后, 重新测定样品的紫外吸收光谱, 实验重现性好.

图2 4-DHBTU和4-AHBT在CH3CN溶液中的紫外吸收光谱Fig.2 UV absorption spectra of 4-DHBTU and 4-AHBT in CH3CN solution
为了反映光学性能的改进, 图2同时给出了相同条件下4-AHBT的紫外光谱. 可以看出, 4-DHBTU的紫外吸收强度显著提高, 约为4-AHBT的 3.5倍, 最大吸收峰位红移明显, 由347 nm位移到367nm, 红移20 nm; 有趣的是, 与4-AHBT不同, 在4-DHBTU的紫外吸收谱图中, 出现了两个强吸收峰.由于紫外吸收光谱强度、最大吸收峰位等与分子的共轭结构有着密切的联系, 因此, 容易推断4-DHBTU分子共轭结构区域较4-AHBT分子的成倍增加, 是导致4-DHBTU紫外最大吸收峰红移的可能原因. 为了进一步验证红移原因, 同时对其双峰结构给予合理的、定量的解释, 我们将在下面结合密度泛函理论计算结果进行讨论.

图3 4-DHBTU 在基态时的6种醇式异构体及相对能量Fig.3 Six enol isomers and their relative energies of 4-DHBTU in the ground state
2.2.3 4-DHBTU的密度泛函理论计算
利用密度泛函理论方法, 在B3LYP/6-31+G*水平下, 对由2个4-AHBT与1个羰基反应生成的6种可能的4-DHBTU基态结构进行了几何全优化(见图3),然后,在同一理论水平下, 计算了优化结构的IR光谱和氢谱NMR化学位移. 为解析实验紫外光谱, 采用TD-DFT方法, 在同一计算水平下, 计算了4-DHBTU分子的垂直跃迁能. 为了考察溶剂效应的影响, 本文中除特别说明的情况外, 均采用连续介质模型(PCM)23进行几何结构优化, 然后再在相同的理论水平下计算光谱. 全部计算均是利用Gaussian 03程序包24完成的. 需要说明的是, 为了确定泛函与基组选取的合理性, 我们测试了在4种泛函(B3LYP, PBEPBE, BPV86, MPW1PW91), 2种基组(6-31+G*, 6-311+G**)下, 4-DHBTU的优化几何结构及其振动频率(计算结果见Supporting Information中的表S1–S2),并与实验IR结果进行比较. 计算表明, B3LYP/6-31+G*计算水平适合于4-DHBTU分子. 这与袁彦杰等25在研究4-AHBT单体几何结构与光谱性质时证明的B3LYP/6-31+G*计算水平可以得到与实验结果符合较好的结论是一致的.
3 结果与讨论
3.1 4-DHBTU的几何结构优化
4-DHBTU分子是由两个4-AHBT结构单元(单体)通过一个平面的脲结构连接而成. 图1可以看出,由稳定的4-AHBT结构出发, 能够得到trans与cis两种4-DHBTU分子异构体, 即trans-4-DHBTU (简写为trans-C11)与cis-4-DHBTU (简写为cis-C11). 值得关注的是, 由于trans与cis异构体转化需要大量能量以破坏π电子的共轭体系, 因此, 在合成过程中, 二者并不能自由地相互转换. 也就是说, 4-DHBTU异构体中仅有羟基氢可以自由转动, 形成分子内或分子间氢键. 考虑到溶液环境中, 4-DHBTU分子与溶剂分子的氢键作用可能导致部分的4-DHBTU分子内氢键破环, 因此, 液态环境中4-DHBTU分子可能存在的稳定构象为6种醇式异构体, 包括C11, C22, C12的 cis与trans异构体, 见图3.
在B3LYP/6-31+G*水平下, 对这6种醇式异构体的几何结构进行全优化, 结果表明, C11型异构体为平面结构, C22、C12型异构体为非平面结构, 在能量方面, 由于苯环上的羟基H与噻唑环上的N形成氢键, 因此, C11型4-DHBTU能量最低, 最稳定; 而没有形成氢键的C22型4-DHBTU能量最高; 具有1个分子内氢键的C12能量则介于两者之间. 此外, 计算表明每种结构的cis与trans异构体能量相近.
3.2 4-DHBTU分子的红外和核磁共振光谱
为了考察实验样本4-DHBTU中可能存在的异构体情况, 利用密度泛函理论方法, 在B3LYP/6-31+G*水平下, 计算了基态下6种醇式4-DHBTU异构体的IR光谱以及NMR氢谱, 并与实验光谱进行了比较分析. 由于顺反异构体对计算IR光谱影响不大,因此, 除基态C11构象外, 表1仅给出C12、C22反式构象的振动频率; 此外, 理论计算可以得到3N – 6, 即156个振动模式, 并能够容易地借助Gaussview等图形软件对各个振动模式进行分类归属, 然而, 由于4-DHBTU分子为一个大的共轭体系, 实验红外谱图中可以明确指认的谱峰有限(主要是与伸缩振动相关谱峰), 为了便于与实验谱图进行比较分析, 表1中我们只列出了指纹区外14个可以归属的振动区域, 涉及所有单键、双键以及离域共轭伸缩振动模式.

表1 4-DHBTU理论计算和实验红外光谱数据及其归属Table1 Calculated and observed IR spectral data of 4-DHBTU with band assignments
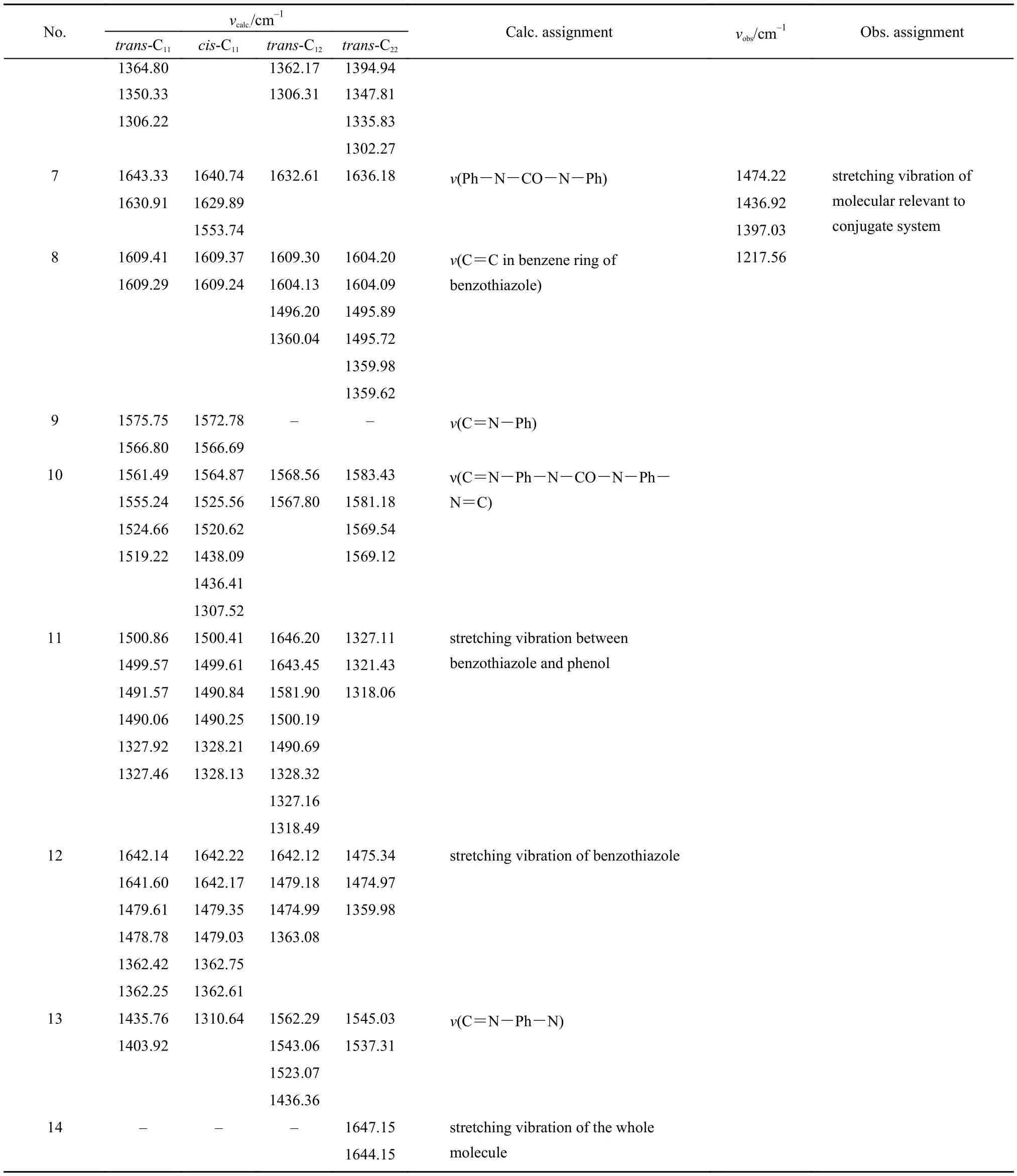
continued Table 1
表1中值得一提的是O-H键的伸缩振动模式.理论计算表明, 由于C11的顺反异构体中羟基各形成2个分子内氢键, 其O-H振动频率均约为3300 cm–1,与我们实验光谱中的羟基振动频率(3275.65 cm–1)符合较好, 其值也与凝聚态中水的伸缩振动频率相近;异构体trans-C12的2个羟基, 其中一个形成分子内氢键, 另外一个为自由悬挂式羟基, 振动模式分别为表1中No.2和No.3, 后者的振动频率较前者小约500 cm–1; 而异构体trans-C22中2个羟基均为自由悬挂式羟基, 其振动频率值约为3750 cm–1, 这与气相中水分子的伸缩振动频率相近.
由于4-DHBTU样品的难溶性, 实验光谱所测为固体样品. 对比理论计算与实验观测羟基振动模式,实验光谱表明观测样品不存在自由羟基振动模式,其羟基均形成氢键, 但不排除分子间氢键的可能性.由于本文中理论模型只考虑了气相单分子情况, 不能考察分子间可能存在的氢键作用, 因而, 仅凭实验光谱还不能排除trans-C12和trans-C22在4-DHBTU样品中存在的可能性. 为了进一步考察4-DHBTU样品中可能存在的异构体组成信息, 我们进一步在理论计算与实验观测两方面研究了4-DHBTU分子的1H NMR谱.
表2列出了4-DHBTU分子的理论1H NMR化学位移, 考虑到实验中样品4-DHBTU的核磁测定是在DMSO溶剂中进行的, 为了更好地对理论计算与实验1H NMR化学位移进行比较, 还利用隐式连续介质模型(PCM)计算了C11顺反两种异构体, 以及异构体trans-C12、trans-C22的溶剂效应影响. 为方便讨论,根据溶剂效应对1H NMR化学位移的影响, 我们把4-DHBTU分子中18个氢原子分为3组: 噻唑环上的碳氢(1H–8H); 苯环上的碳氢(9H–14H); 以及可作为氢键给体的氮氢(15H, 16H)和氧氢(17H, 18H). 容易看出, 在真空条件下, 6种构象的4-DHBTU分子中, 噻唑环上8个碳氢原子的化学位移略小于实验观测值(偏差小于0.3), PCM溶剂校正可以使化学位移增大约0.2, 但总体上讲, 溶剂效应影响不大; 而苯环上6个碳氢原子中, 虽然有部分碳氢原子的化学位移与实验观测值有较大差异(约为2), 但通过简单的PCM溶剂校正, 这些原子的位移计算值与实验值的差值均可小于1; 与前两组碳氢原子不同, 氮氢的化学位移值与实验值相差较大, 约为4, 而PCM溶剂校正只能增大1. 考虑到15H、16H与DMSO中的氧原子可能的氢键作用, 以cis-C11为例, 我们尝试设计了显式溶质–溶剂相互作用的簇合物模型计算溶剂效应, 如图4所示. 计算结果显示, 在簇合物溶剂模型下, 计算值与实验值符合得相当好, 说明对于较强的溶剂化效应, 如氢键作用时, 溶剂模型选择是非常重要的. 此外, 氧氢的情况可分为两种, 一种是形成分子内氢键的, 如异构体trans-C11与cis-C11中的17H、18H以及trans-C12、cis-C12中的17H, 它们的计算氢谱位移与实验值较接近, 误差小于1, 溶剂效应影响不大; 另一种未形成氢键的, 如与trans-C22、cis-C22中的17H、18H以及trans-C12、cis-C12中的18H, 与氮氢的情况类似. 在显式的簇合物溶剂化模型下, 溶质与溶剂DMSO间的氢键作用使得氢谱位移显著增大, 并趋近于实验值.
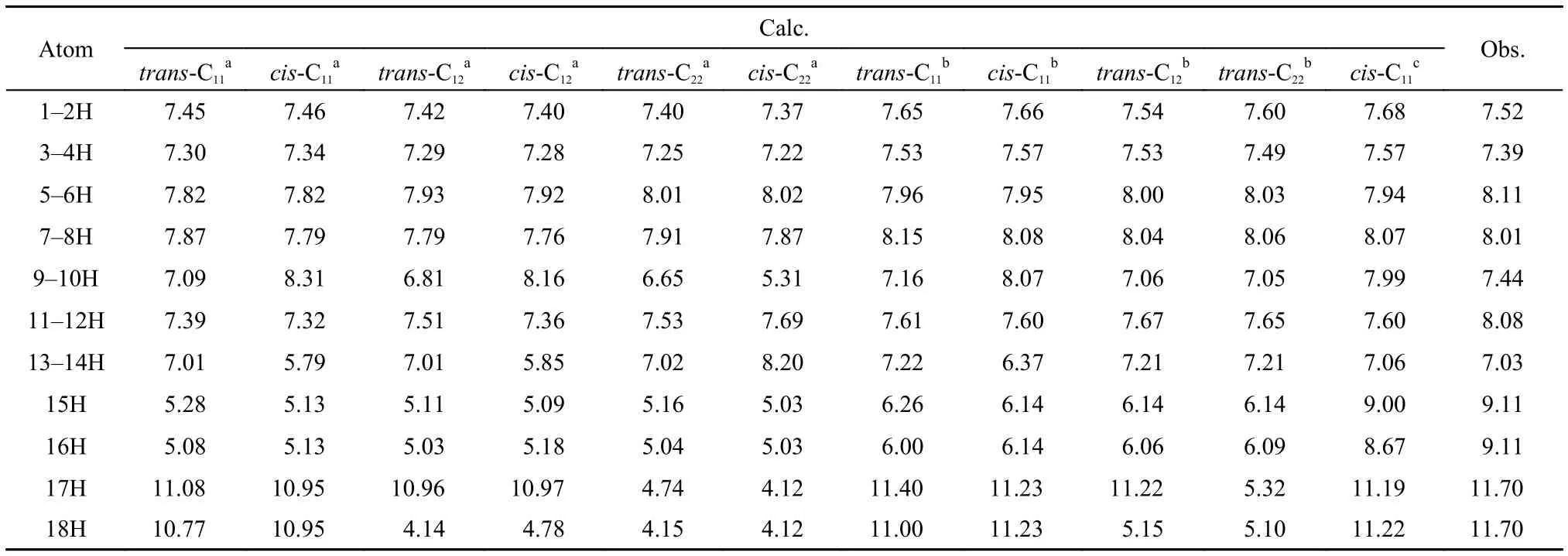
表2 4-DHBTU理论计算和实验氢谱核磁共振化学位移Table2 Calculated and observed1H NMR chemical shifts of 4-DHBTU
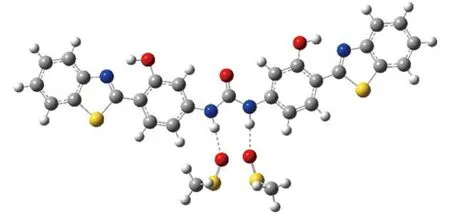
图4 溶剂化的簇模型Fig.4 Cluster model for solvation
上述结果显示, 虽然在真空下, 6种4-DHBTU醇式异构体分子的氢谱位移与实验值具有较大偏差,但经溶剂校正后, 它们的计算值均与实验值相近,这表明氢谱核磁位移结果不能排除在DMSO溶剂中多种4-DHBTU分子异构体共存的可能性.
3.3 4-DHBTU的紫外光谱
与单体4-AHBT相比, 实验观测的4-DHBTU紫外吸收强度显著增强, 最大吸收峰发生了明显红移,并呈现出双吸收峰特征. 为了合理地解释上述特性,我们尝试利用密度泛函理论方法, 在B3LYP/6-31+G*水平下, 计算了4-DHBTU分子的垂直跃迁能,得到了4个异构体的理论吸收光谱数据(见表3). 可以看出, 从最稳定异构体trans-C11与cis-C11的基态到第一激发态(S1)的垂直跃迁能分别为3.39 eV (365.23nm) 与3.41 eV (363.64 nm), 与我们的实验观测值3.38 eV (367 nm)非常吻合; S1态的谐振强度(f = 1.78, 1.80)是最大的, 表明S1态是构型trans-C11与cis-C11的最强吸收态, 对应于实验观测的最大吸收峰.为考虑溶剂效应的影响, 我们采用PCM计算了trans-C11与cis-C11在乙腈中的吸收光谱. 计算结果显示, 从基态到第一激发态(S1)的垂直跃迁能分别为3.30 eV (375.21 nm)与3.31 eV (374.78 nm), S1态的谐振强度f均为2.09, 可见, 溶剂对第一激发态的垂直跃迁能(红移约10 nm)影响不大.
接下来, 归属光谱双峰中的次强吸收峰, 首先排除了该吸收峰为trans-C11或cis-C11异构体从基态到S2态的吸收跃迁的可能性. 计算显示, trans-C11或cis-C11基态到S2态的吸收能, 约为3.70 eV (335.17nm) 处或3.69 eV (335.83 nm) , 但其谐振强度很小(f = 0.04或f = 0.01), 紫外光谱图中没有观察到对应的弱吸收峰. 此外, 通过质谱实验验证了紫外光谱样品中不包含4-AHBT, 进一步排除了该峰为4-AHBT吸收峰的可能性. 那么, 次强吸收峰是其他异构体的第一激发态的跃迁吸收吗?
为此, 我们分析了表3中其他4-DHBTU异构体的垂直激发能. 可以看出, trans-C22与cis-C22的S0→S1激发能分别为3.66 eV (338.43 nm, f = 1.74)与 3.65 eV (339.73 nm, f = 1.71), 与紫外光谱中的次强吸收峰峰位3.54 eV (350 nm)相差约10–11 nm. 若考虑溶剂效应, trans-C22与cis-C22的S1激发能分别3.53eV (351.36 nm, f = 2.01)与3.53 eV (351.31 nm, f = 1.97),与紫外光谱中的次强吸收峰峰位非常接近. 由此,我们有理由推测次强吸收峰是由cis-C22与trans-C22基态激发产生的. 从能量角度来看, 异构体C22能量较C11能量高约1 eV, C22的形成需要C11在溶剂环境下克服分子内氢键得到, 异构体C22存在的概率较C11小, 一定程度说明了异构体C22跃迁强度较异构体C11跃迁强度弱的原因. 但在极性溶液环境中C22分子中自由的OH键, 不仅可以通过热转动, 向低能的异构体C11转化(分子内氢键), 同时, 也可以与溶剂或其他溶质分子形成分子间氢键, 使其能量降低, 从而稳定存在. 此外, 类似的分析表明, 交叉式的异构体cis-C12和trans-C12的垂直跃迁能对应的吸收也可能包含在紫外吸收的大峰之中. 也就是说, cis-C11、trans-C11与cis-C22、trans-C22等多种异构体的共同存在是导致4-DHBTU紫外光谱变宽并产生双峰的主要原因.
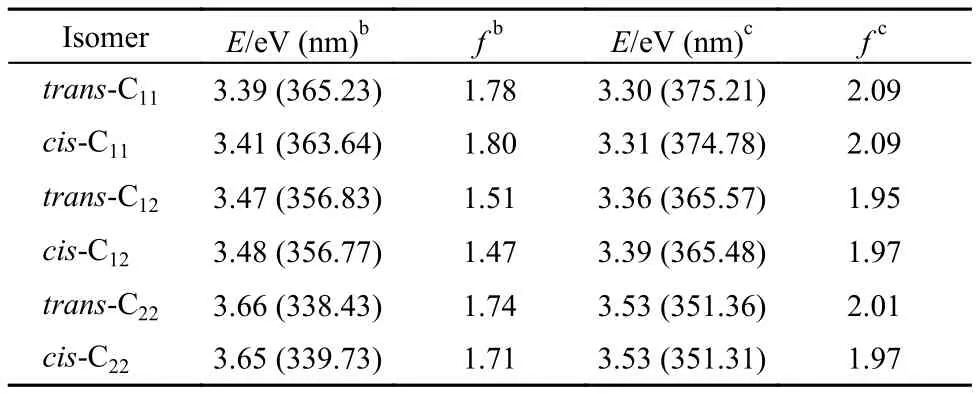
表3 4-DHBTU的S0→S1态激发能aTable3 Excitation energiesa(S0→S1) of 4-DHBTU
4 结 论
由两个4-AHBT结构单元组成的新的化合物4-DHBTU, 其紫外吸收强度比4-AHBT显著增强, 最大吸收峰发生了明显红移, 并出现了双强吸收峰.
采用密度泛函理论方法, 在B3LYP/6-31+G*水平上优化得到了6种可能的4-DHBTU分子基态的稳定异构体, 计算了其振动频率以及氢谱核磁共振化学位移, 在此基础上, 利用含时密度泛函理论, 在同一水平上计算了4-DHBTU分子垂直跃迁能. 结果表明, 理论计算与实验光谱较好地相符合, 溶剂DMSO与4-DHBTU分子氢键作用是4-DHBTU分子15H、16H 氢谱NMR化学位移增大的主要原因, 而trans-C11、cis-C11与trans-C22、cis-C22等多种异构体的共存是导致4-DHBTU紫外光谱变宽并产生双强峰的主要原因.
Supporting Information: The optimized geometries of trans-C11and vibration frequencies at 6-31+G* and 6-311++G** basis sets with four functionals of B3LYP, PBEPBE, BPV86, and MPW1PW91 are included. This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1)Ma, D.; Liang, F. S.; Wang, L. X.; Lee, S. T.; Hung, L. S. Chem. Phys. Lett. 2002, 358, 24. doi: 10.1016/S0009-2614(02)00546-8
(2)Rodembusch, F. S.; Tiago, B.; Segala, M.; Tavares, L.; Correia, R. R. B.; Stefani, V. Chem. Phys. 2004, 305, 115. doi: 10.1016/j.chemphys.2004.06.046
(3)Tolbert, L. M.; Solntsev, K. M. Accounts Chem. Res. 2002, 35, 19. doi: 10.1021/ar990109f
(4)Ma, Q. J.; Zhang, X. B.; Zhao, X. H.; Gong, Y. J.; Tang, J.; Shen, G. L.; Yu, R. Q. Spectrochim. Acta Part A 2009, 73, 687. doi: 10.1016/j.saa.2009.03.023
(5)Henary, M. M.; Fahrni, J. C. J. Phys. Chem. A 2002, 106, 5210. doi: 10.1021/jp014634j
(6)Zhang, X. B.; Peng, J.; He, C. L.; Shen, G. L.; Yu, R. Q. Anal. Chim. Acta 2006, 567, 189. doi: 10.1016/j.aca. 2006.03.025
(7)Qin, W.; Obare, S. O.; Murphy, C. J.; Angel, S. M. Anal. Chem. 2002, 74, 4757. doi: 10.1021/ac020365x
(8)Purkayastha, P.; Chattopadhyay, N. Int. J. Mol. Sci. 2003, 4, 335. doi: 10.3390/i4060335
(9)Chipem, F. A. S.; Krishnamoorthy, G. J. Phys. Chem. B 2013, 117, 14079. doi: 10.1021/jp405804c
(10)Luber, S.; Adamczyk, K.; Nibbering, E. T. J.; Batista, V. S. J. Phys. Chem. A 2013, 117, 5269. doi: 10.1021/jp403342w
(11)Benelhadj, K.; Massue, J.; Retailleau, P.; Ulrich, G.; Ziessel R. Org. Lett. 2013, 15, 2918.
(12)Padalkar, V. S.; Tathe, A. B.; Gupta, V. D.; Patil, V. S.; Phatangare, K.; Sekar, N. J. Fluoresc. 2012, 22, 311. doi: 10.1007/s10895-011-0962-8
(13)Patil, V. S.; Padalkar, V. S.; Tathe, A. B.; Gupta, V. D.; Sekar, N. J. Fluoresc. 2013, 23, 1019.
(14)Zhao, J. Z.; Ji, S. M.; Chen, Y. H.; Guo, H. M.; Yang, P. Phys. Chem. Chem. Phys. 2012, 14, 8803. doi: 10.1039/c2cp23144a
(15)Abou-Zied, O. K. Phys. Chem. Chem. Phys. 2012, 14, 2832. doi: 10.1039/c2cp23337a
(16)Abou-Zied, O. K. RSC Adv. 2013, 3, 8747. doi: 10.1039/c3ra40907d
(17)Syetov, Y. J. Fluoresc. 2013, 23, 689. doi: 10.1007/ s10895-013-1196-8
(18)Rodembusch, F. S.; Leusin, F. P.; Campo, L. F.; Stefani, V. J. Lumin. 2007, 126, 728. doi: 10.1016/j.jlumin. 2006.11.007
(19)Wu, Y. F.; Cui, Y. N.; Li, S. M.; Jia, Y. P.; Yin, J. M. Chin. J. Inorg. Chem. 2012, 28, 910. [吴玉防, 崔颖娜, 李慎敏, 贾颖萍,尹静梅. 无机化学学报, 2012, 28, 910.]
(20)Yin, J. M.; Yang, Q.; Jia, Y. P.; Cui, Y, N.; Zhou, G. Y.; Gao, D. B. Synthesis and Application of N,N’-di[3-hydroxy-4-(2-benzothiazole)phenyl]urea. CN Patent ZL200910012518.6, 2011-05-25. [尹静梅, 阳 强, 贾颖萍, 崔颖娜, 周广运, 高大彬.一种N,N’-二-[3-羟基-4-(2-苯并噻唑)苯基]脲的合成方法及应用, 中国, ZL200910012518.6[P], 2011-05-25.]
(21)Stratmann, R. E.; Scuseria, G. E.; Frisch, M. J. J. Chem. Phys. 1998, 109, 8218. doi: 10.1063/1.477483
(22)Bauernschmitt, R.; Ahlrichs, R. Chem. Phys. Lett. 1996, 256, 454. doi: 10.1016/0009-2614(96)00440-X
(23)Peng, H. L.; Yu, X. Y.; Yi, P. G.; Wang, Z. X.; Li, X. F.; Wang, T.; Zhou, J. M. Acta Phys. -Chim. Sin. 2010, 26, 141. [彭洪亮,于贤勇, 易平贵, 汪朝旭, 李筱芳, 王 涛, 周继明. 物理化学学报, 2010, 26, 141.] doi: 10.3866/PKU.WHXB 20100130
(24)Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 03, Revision B.04; Gaussian Inc.: Pittsburgh, PA, 2003.
(25)Yuan, Y. J.; He, R. X.; Hu, G. J.; Li, M. Journal of Southwest University (Natural Science Edition) 2009, 31, 55. [袁彦杰, 何荣幸, 胡广剑, 李 明. 西南大学学报(自然科学版), 2009, 31, 55.]
Experimental and Density Functional Theoretical Studies on the Spectra of N,N′-di[3-hydroxy-4-(2-benzothiazole)phenyl]urea
CUI Ying-Na1,*YIN Jing-Mei1,2JIA Ying-Ping1LI Shen-Min1,2,*
(1College of Environment and Chemical Engineering, Dalian University, Dalian 116622, Liaoning Province, P. R. China;2Liaoning Key Laboratory of Bio-organic Chemistry, Dalian University, Dalian 116622, Liaoning Province, P. R. China)
The experimental infrared (IR), nuclear magnetic resonance (NMR), and ultraviolet (UV) spectra, and density functional theory (DFT) calculations of the novel compound N,N′-di[3-hydroxy-4-(2-benzothiazole) phenyl]urea (4-DHBTU) are presented. Compared with the UV spectra of the 2-(4-amino-2-hydroxyphenyl) benzothiazole (4-AHBT) monomer, the experimental spectra of 4-DHBTU, a dimer of 4-AHBT, show dualwavelength absorption with significantly enhanced absorption intensity and an obvious red shift of the maximum absorption peak. Analysis of the experimental spectra and the DFT calculations shows that the structures of cis-C11and trans-C11are the two most stable conformers, and that the main reason for the different UV spectral properties of the dimer and monomer is the coexistence of cis-C11, trans-C11, cis-C22, and trans-C22in the 4-DHBTU sample. In addition, the DFT calculations indicate that a hydrogen-bonding interaction between 4-DHBTU and the dimethyl sulfoxide (DMSO) solvent leads to a large1H NMR chemical shift for atoms 15H and 16H in 4-DHBTU.
N,N′-di[3-hydroxy-4-(2-benzothiazole)phenyl]urea; Ultraviolet spectroscopy; Excited state; Density functional theory
O641
10.3866/PKU.WHXB201506182
Received: May 11, 2015; Revised: June 17, 2015; Published on Web: June 18, 2015.
*Corresponding authors. LI Shen-Min, Email: lishenmin@dlu.edu.cn; Tel: +86-411-87403949. CUI Ying-Na, Email: cuiyn2008@163.com;
Tel: +86-411-87402434.
The project was supported by the National Natural Science Foundation of China (21133005), National Natural Science Foundation of Liaoning Province, China (201205535), and Scientific Research Foundation of the Higher Education Institutions of Education Bureau of Liaoning Province (Key Laboratory Project 2010), China (LS2010005).
国家自然科学基金(21133005), 辽宁省自然科学基金(201205535)和辽宁省教育厅2010年度高等学校科研项目(重点实验室项目) (LS2010005)资助
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
食品科学技术学报(2022年6期)2022-12-15
现代临床医学(2022年4期)2022-09-29
中国抗生素杂志(2022年7期)2022-08-18
化工与医药工程(2022年3期)2022-08-08
浙江化工(2022年4期)2022-05-07
科学技术创新(2022年36期)2022-02-01
中国油脂(2019年1期)2019-01-23
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
