
金属串配合物[Ni3(L)4(NCS)2](L = dpa–, mpta–, mdpa–, mppa–)结构和磁性的理论研究
2015-12-05 06:29周沃华吴子文徐志广
物理化学学报 2015年9期
陈 蓉 周沃华 吴子文 许 旋,2,3,* 徐志广
(1华南师范大学化学与环境学院, 广州 510006; 2教育部环境理论化学重点实验室, 广州 510006;3广州市能源转化与储能材料重点实验室, 广州 510006)
金属串配合物[Ni3(L)4(NCS)2](L = dpa–, mpta–, mdpa–, mppa–)结构和磁性的理论研究
陈 蓉1周沃华1吴子文1许 旋1,2,3,*徐志广1
(1华南师范大学化学与环境学院, 广州 510006;2教育部环境理论化学重点实验室, 广州 510006;3广州市能源转化与储能材料重点实验室, 广州 510006)
应用密度泛函理论BP86方法结合自然键轨道分析方法对具有分子导线潜在应用前景的金属串配合物[Ni3(L)4(NCS)2](L = dpa–(1), mpta–(2), mdpa–(3), mppa–(4))进行研究, 分析了桥联配体L对Ni―Ni相互作用和磁耦合性质的影响. 结果得到: (1) 配合物的基态均是对应于五重态(HS)的反铁磁(AF)单重态, HS的能量和结构与AF态相近,链形成了三中心四电子引入甲基成为mdpa–, 对Ni―Ni、 Ni―N距离影响不大; 3H-吡咯环和噻唑环取代吡啶环后, N1―N2、Ni―Ni距离增大, Ni2―N2键长缩短, 但噻唑环的影响较小; 故Ni―Ni相互作用强度为1 ≈ 3 > 2 > 4. (3) 预测了3和4的Jab值为–103和–88 cm–1, 随 Ni―Ni相互作用增强磁耦合效应增大. Ni―Ni相互作用越大, 通过链σ型轨道的直接磁耦合越强; Ni2―N2键越强, 通过涉及桥联配体的间接磁耦合越强, 直接磁耦合比间接磁耦合更强.
金属串配合物; 桥联配体; 密度泛函理论; 磁耦合; 分子导线
1 引 言
由于受常规微型化硅器件的限制, 分子器件的发展备受关注,1–6而线性排列的金属串配合物有望成为"分子导线"或是"纳米导线"材料. 1968年首次制备的第一个金属串配合物[Ni3(dpa)4Cl2]7(dpa–为二吡啶胺)于1990年进行单晶结构测定和分析,8发现Ni原子和轴向配体Cl–呈直线排列, 四个桥联配体dpa–螺旋盘绕着金属轴并与三个Ni原子配位, 基态为反铁磁(AF)单重态, 两端的Ni原子为磁性中心, 各有两个单电子. Peng等9–11用扫描隧道显微(STM)对Ni3(dpa)4(NCS)2(1)的研究发现其导电性较好, 电导和磁耦合常数分别为3.8×10–3G0(G0= 2e2/h ≈ 12.9 kΩ–1)和–122 cm–1; M―M相互作用越强, 电荷转移效率越高, 导电能力越强. Cotton等12,13早期的理论研究根据Ni―Ni键长和Ni的d8价电子结构认为未形成Ni―Ni键, 为抗磁性, 与实验结果不符. 2004年, Bénard等14用密度泛函理论(DFT)计算了Ni3(dpa)4Cl2的电子结构和磁性, 认为存在Ni―Ni相互作用, 较好地解释了AF耦合现象.

图1 配合物1–4的结构Fig.1 Structures of complexes 1–4
桥联配体的结构是影响M―M相互作用和电荷转移性质的主要因素之一. Cotton等15用HPhPyF配体(N,N-苯基吡啶基甲脒)取代dpa–发现可减小吡啶环上H原子间的排斥作用; Cotton等16用互扣的两个半包围配体mpeptea2–(N,N'-bis[(6'-pyrid-2''-yl)amin-opyrid-2'-yl] bismethyl-2,6-diaminopyridine)代替四个dpa–配体和两个轴向配体, 解决了轴向配体与端位Ni结合的不稳定性; Peng等17将dpa–改为dpza–(二吡嗪胺), 提高了配合物的氧化还原稳定性. 最近, Peng等18还合成了含非对称的mpta–配体(4-甲基吡啶基噻唑胺)的Ni3(mpta)4(NCS)2(2)(见图1), 由晶体结构发现Ni―Ni键长(0.2471 nm)大于1的0.2429 nm, AF耦合(–91 cm–1)和导电性(1.8 × 10–3G0)均比1的弱.相对于dpa–, mpta–左边的吡啶环多了一个甲基, 右边的吡啶环改为五元噻唑环. 为分析桥联配体L结构变化对配合物结构和性质的影响, 设计了含4-甲基-2,2'-二吡啶胺(mdpa–)的配合物(3)、4-甲基吡啶基-3H-吡咯胺(mppa–)的配合物(4), 应用DFT方法对配合物1–4(见图1)的结构及其磁性进行研究, 为设计合成新的金属串配合物及其分子器件的应用提供理论参考.

表1 配合物1–4优化的分子能量(a.u.)Table1 Optimized molecular energies (a.u.) for complexes 1–4
2 计算方法
Ni(II)的组态是3d8, 故考虑配合物各种可能的自旋态: 闭壳层单重态(CS)、三重态、五重态(HS)、七重态. 采用密度泛函理论(DFT)的B3LYP19,20和BP8621,22方法, C、N和H用6-31G*基组, S用6-311G*基组, Ni分别用SDD23基组(a)和LANL2DZ24基组(b), 对配合物1–4及其相应的桥联配体进行几何优化. 对优化构型(如图2)进行振动频率分析, 发现均无虚频, 并进行自然键轨道(NBO)25分析. 采用对称性破损理论(BS)方法26计算其AF单重态, 并根据Yamaguchi等27提出自旋投影结合铁磁耦合态(FM)由式(1)计算磁耦合交换常数Jab值:

上式中EX和
3 结果与讨论
3.1 配合物的稳定性和几何结构
由表1可见各种方法计算的配合物1–4的HS能量均远低于CS、三重态和七重态. 对应于HS的AF单重态的能量略低于HS (0.0016–0.0020 a.u.), 表明基态为AF态, 与磁性实验得到为两端Ni各有两个单电子的AF态的结果相符. 由表2可见, 采用BP86方法和SDD基组计算的1的结构最接近实验值, 故选用BP86方法SDD基组对计算结果进行分析.
HS态的几何参数与AF态的接近且规律相似. AF态下, 配合物1–4的Ni―Ni键和桥联配位Ni―N键的Wiberg键级分别为0.2668–0.2948和0.3218– 0.4223, 表明存在弱的Ni―Ni、Ni―N相互作用. 由于胺基N1的碱性较端位N2、N3的强, 故中间的Ni1―N1键长(0.1919–0.1936 nm)较端位Ni2―N2、Ni3―N3的键长(0.2047–0.2104 nm)短, 两端的Ni―N键较弱. 由表3发现, 桥联配体的改变影响Ni―Ni、Ni2―N2键长, 对于Ni―Ni键长1 (0.2431nm) ≈ 3 (0.2432 nm) < 2 (0.2485 nm) < 4 (0.2489 nm), 而Ni2―N2键长1 (0.2095 nm) ≈ 3 (0.2097 nm)>2 (0.2074 nm)>4 (0.2047 nm). 3与1的Ni―Ni、Ni―N键长差异很小(0.0001–0.0005 nm), 表明mdpa–中吡啶环上甲基的引入对Ni―Ni、Ni―N键影响不大. 4的Ni―Ni键长比3的显著增大了0.0057nm, Ni2―N2键长却比3的缩短了0.0050 nm, 表明mdpa–的六元吡啶环被五元的3H-吡咯环取代形成mppa–后Ni―N键增强, 而Ni―Ni键被削弱. 当4的3H-吡咯环变成2中mpta–的五元噻唑环后, Ni2―N2键长略为增长, 而Ni―Ni键长略为缩短.

表2 配合物1的部分键长(d)和二面角(A)Table2 Selected bond lengths (d) and dihedral angles (A) for complex 1

表3 采用BP86泛函计算配合物1–4的部分几何参数、S2和自旋密度Table3 Selected geometrical parameters, S2, and spin densities calculated for complexes 1–4 by using BP86 functional
3.2 分子电子组态和Ni―Ni成键分析
Ni2+为d8结构,链中由组成的δ轨道主要用于与桥联配体L的N原子结合; 其余d轨道组成12个包括σ、π和δ类的分子轨道. 若这12个分子轨道填充24个d电子则不存在Ni―Ni相互作用, 应为抗磁性, 这与基态为AF单重态的实验现象18不符. 由图3的分子轨道能级图和图4的分子轨道图可见, 由于σnb、σ*轨道中轴向配体NCS中N的2p轨道与Ni的反键作用, 使σnb、σ*轨道能升高; 此外, 桥联配体上碱性较弱的N2、N3与Ni2、Ni3形成的Ni―N键较弱,使两个轨道(Ni的与N的2p轨道组成的σ*)能降低. 因此, σnb、σ*轨道中各有一个电子分别填到了两个轨道上, 形成了σnb、σ*、1和2共4个单占轨道(SOMO). 电子组态均为由图4可见, 1–4中均存在轨道组成的遍及链的σ轨道, 故链形成三中心四电子σ键这与配合物的磁性实验结果和的理论计算结果相符. 此外, σ轨道与σ*轨道轨道能级差大小为1 (2.37 eV) ≈ 3 (2.35 eV) > 2 (2.16 eV) > 4 (2.05 eV), 故Ni―Ni相互作用强度为1 ≈ 3 > 2 > 4.
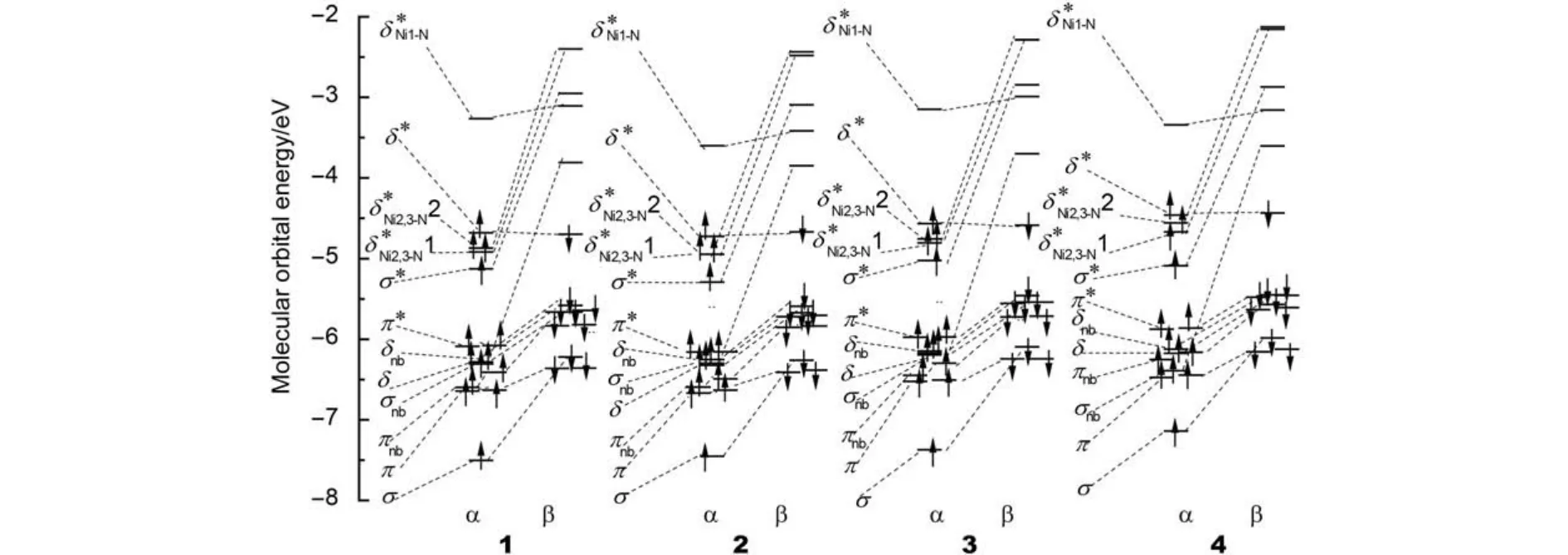
图3 HS态配合物1–4的金属特征轨道能级图Fig.3 Metallic character orbital energy diagram for complexes 1–4 in the HS state
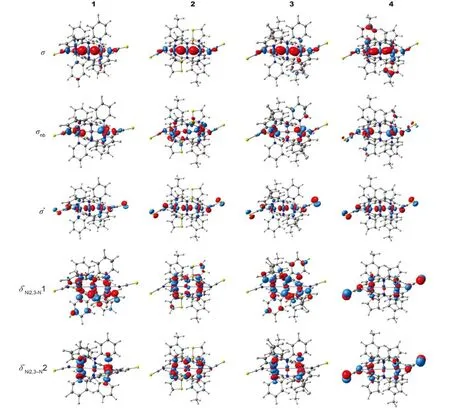
图4 HS态下配合物1–4中的分子轨道图Fig.4 Molecular orbitals diagrams for complexes 1–4 in the HS state
为研究不同桥联配体对Ni―Ni键的影响, 用NBO结果进行分析. 4中3H-吡咯环的C1―N2键中C1为sp2.40杂化, p轨道成分大于3中C1(sp2.29), 因此, 如图1所示, 五元环内角比六元环小, 4的∠C5C1N2 = 110°远小于3的∠C5C1N2 = 120°; 相反, 五元环外N1―C1键中C1为sp2.12杂化, 接近sp2杂化, 其p成分小于3的sp2.24, 使4的角度∠N1C1N2 = 120°大于3的∠N1C1N2 = 116°, 故4中桥联配体mppa–的N2―N3距离0.4688 nm长于3中mdpa–的0.4656 nm,这是五元3H-吡咯环取代吡啶环后Ni―Ni键增长的主要原因. 4的五元3H-吡咯环变成2的五元噻唑环后, 因相同的原因使键角∠N1C1N2进一步增大约2°, 但由于mpta–中S―C1键较弱、键长较长, 使相邻的C1―N1键(0.1320 nm)和C1―N2键(0.1335 nm)比mppa–的0.1338和0.1351 nm短, 故mpta–的N1―N2键长(0.2309 nm)小于mppa–的0.2326 nm, 因此, 2中的Ni―Ni键比4的稍强. 此外, 2和4中桥联配体上五元环的N2原子的负电荷密度–0.520e和–0.569e大于3中N2的–0.494e, 也使2和4的Ni2―N2键比3的强,相反, Ni1―Ni2键则削弱.

图5 HS (a)和AF (b)态配合物1的自旋密度等值线图Fig.5 Contour line map of spin density of complex 1 in the HS (a) and AF (b) states
3.3 配合物的自旋密度和磁耦合
配合物1–4的自旋密度分布相似, 故图5仅列出1的自旋密度等值线图. 两种最低能态均存在四个单电子, 基态AF态中一端Ni(II)的两个单电子自旋向上, 另一端的自旋向下, 自旋密度为±1.435 –±1.438. 能量稍高的HS中两端Ni(II)的四个单电子均自旋向上, 自旋密度为+1.466 – +1.484, 两个磁中心Ni(II)为铁磁耦合, 两端磁中心的自旋单电子向中间的Ni1有一定的离域, Ni1的自旋密度在+0.201 –+0.226范围. AF态和HS态的能量和几何结构都非常接近, 表明AF态和HS态为竞争态, 化学键受自旋耦合影响较小, 这与Bénard等14对Ni3(dpa)4Cl2的研究结果一致. 自旋密度大小顺序为1 ≈ 3 > 2 > 4, 与Ni―Ni相互作用强度顺序一致.
配合物1–4计算的耦合常数Jab分别为–107、–94、–103和–88 cm–1, 1和2的计算值与实验值Jab=–122 cm–1和Jab= –91 cm–1很吻合.10,18耦合强度1 ≈ 3 > 2 > 4, 表明 Ni―Ni键越强, 磁中心Ni(II)的自旋密度越大, 磁中心之间的AF相互作用和自旋极化越强.
López等31对Ni3(dpa)4Cl2的研究认为, 四个单占轨道对两端金属原子的磁相互作用起两种主要作用: (1) 通过沿金属链的σ相互作用的直接磁耦合; (2) 通过涉及金属轨道和桥联配体的δ电子的间接磁耦合. 由图5可见, 自旋密度除主要分布于金属链和轴向配体外, 还有部分在桥联配体的N上, 证实了这两条磁通道的存在. 因链σ的 Ni―Ni相互作用强度为1 ≈ 3 > 2 > 4, 通过金属链σ相互作用的直接磁耦合随Ni―Ni相互作用增强而增大. 而涉及金属轨道和桥联配体的δ电子的间接磁耦合则随桥联配体的N与Ni的相互作用增强而增大, Ni―N键强度差异主要在Ni2―N2, Ni2―N2键强度为4 > 2 > 1 ≈ 3, 故间接磁耦合强度为4 > 2 > 1 ≈ 3.但直接磁耦合效应比间接磁耦合强, 故两种耦合的共同作用导致Jab大小为1 ≈ 3 > 2 > 4.
4 结 论
对[Ni3(L)4(NCS)2]结构的DFT研究得到: (1) 配合物1–4的基态是对应于五重态的AF单重态,链形成了三中心四电子σ键耦合强度随Ni―Ni相互作用增强而增强, 强度次序为1 ≈ 3 > 2 > 4. (3) dpa–的吡啶环上引入甲基对Ni―Ni、Ni―N距离和磁性影响不大; 五元3H-吡咯环和五元噻唑环取代了吡啶环后, 使Ni2―N2键增强, Ni―Ni键减弱, 通过金属链的直接磁耦合减弱, 而通过桥联配体的间接磁耦合增强; 但噻唑环的影响不如3H-吡咯环的影响显著.
(1)Luo, K. G.; Tan, Y.; Xu, X.; Xu, Z. G. Inorg. Chim. Acta 2014, 421, 310. doi: 10.1016/j.ica.2014.06.003
(2)Berry, J. F.; Cotton, F. A.; Murillo, C. A.; Roberts, B. K. Inorg. Chem. 2004, 43, 2277. doi: 10.1021/ic0354320
(3)Chang, H. C.; Li, J. T.; Wang, C. C.; Lin, T. W.; Lee, H. C.; Lee, G. H.; Peng, S. M. Eur. J. Inorg. Chem. 1999, 1999 (8), 1243.
(4)Lai, S. Y.; Wang, C. C.; Chen, Y. H.; Lee, C. C.; Liu, Y. H.; Peng, S. M. J. Chin. Chem. Soc. 1999, 46, 477. doi: 10.1002/jccs.v46.3
(5)Peng, S. M.; Wang, C. C.; Jang, Y. L.; Chen, Y. H.; Li, F. Y.; Mou, C. Y.; Leung, M. K. J. Mag. Mag. Mater. 2000, 209, 80. doi: 10.1016/S0304-8853(99)00650-2
(6)Ismayilov, R. H.;Wang, W. Z.; Lee, G. H.; Yeh, C. Y.; Hua, S. A.; Song, Y.; Rohmer, M. M.; Bénard, M.; Peng, S. M. Angew. Chem. Int. Edit. 2011, 50, 2045. doi: 10.1002/anie.v50.9
(7)Hurley, T. J.; Robinson, M. A. Inorg. Chem. 1968, 7 (1), 33. doi: 10.1021/ic50059a007
(8)Aduldecha, S.; Hathaway, B. J. Chem. Soc. Dalton Trans. 1991, 993.
(9)Lin, S. Y.; Chen, I. W. P.; Chen, C. H.; Hsieh, M. H.; Yeh, C. Y.; Lin, T. W.; Chen, Y. H.; Peng, S. M. J. Phys. Chem. B 2004, 108, 959. doi: 10.1021/jp035415w
(10)Shieh, S. J.; Chou, C. C.; Lee, G. H.; Wang, C. C.; Peng, S. M. Angew. Chem. Int. Edit. 1997, 36, 56.
(11)Cheng, M. C.; Liu, I. P. C.; Hsu, C. H.; Lee, G. H.; Chen, C. H.; Peng, S. M. Dalton Trans. 2012, 41, 3166. doi: 10.1039/c2dt11246a
(12)Clérac, R.; Cotton, F. A.; Dunbar, K. R.; Murillo, C. A.; Pascual, I.; Wang, X. P. Inorg. Chem. 1999, 38, 2655. doi: 10.1021/ic990006t
(13)Berry, J. F.; Cotton, F. A.; Daniels, L. M.; Murillo, C. A.; Wang, X. P. Inorg. Chem. 2003, 42 (7), 2418. doi: 10.1021/ic0262740
(14)Kiehl, P.; Rohmer, M. M.; Bénard, M. Inorg. Chem. 2004, 43 (10), 3151. doi: 10.1021/ic040011j
(15)Cotton, F. A.; Lei, P.; Murillo, C. A. Inorg. Chim. Acta 2003, 351, 183. doi: 10.1016/S0020-1693(03)00112-9
(16)Cotton, F. A.; Chao, H.; Murillo, C. A.; Wang, Q. S. Dalton Trans. 2006, No. 45, 5416.
(17)Ismayilov, R. H.; Wang, W. Z.; Lee, G. H.; Wang, R. R.; Liu, I. P. C.; Yeh, C. Y.; Peng, S. M. Dalton Trans. 2007, 21 (27), 2898.
(18)Yang, C. C.; Liu, I. P. C.; Hsu, Y. J.; Lee, G. H.; Chen, C. H.; Peng, S. M. Eur. J. Inorg. Chem. 2013, 2013 (2), 263. doi: 10.1002/ejic.201200934
(19)Becke, A. D. J. Chem. Phys. 1993, 98, 5648. doi: 10.1063/1.464913
(20)Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785. doi: 10.1103/PhysRevB.37.785
(21)Becke, A. D. Phys. Rev. A 1988, 38 (6), 3098. doi: 10.1103/PhysRevA.38.3098
(22)Perdew, J. P. Phys. Rev. 1986, B33, 8882; 1986, B34, 7406.
(23)Schwerdtfeger, P.; Dolg, M.; Schwarz, W. H. E.; Bowmaker, G. A.; Boyd, P. D. J. Chem. Phys. 1989, 91, 1762. doi: 10.1063/1.457082
(24)Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 299. doi: 10.1063/1.448975
(25)Glendening, E. D.; Reed, A. E.; Carpenter, J. E.; Weinhold, F. NBO, Version 3.1; Theoretical Chemistry Institute, University of Wisconsin: Madison, 1996.
(26)Noodleman, L. J. Chem. Phys. 1981, 74 (10), 5737. doi: 10.1063/1.440939
(27)Kitagawa, Y.; Matsui, T.; Nakanishi, Y.; Shigeta, Y.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Dalton Trans. 2013, 42, 16200. doi: 10.1039/c3dt51466h
(28)Lu, T. Multiwfn, Revision 3.3.5; Beijing Kein Research Center for Natural Sciences: Beijing, 2014.
(29)Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Pittsburgh, PA, 2009.
(30)Tan, Y.; Huang, X.; Xu, X.; Xu, Z. G. Chem. J. Chin. Univ. 2012, 33, 1278. [谭 莹, 黄 晓, 许 旋, 徐志广. 高等学校化学学报, 2012, 33, 1278.]
(31)López, X.; Bénard, M.; Rohmer, M. M. J. Mol. Struct. 2006, 777, 53. doi: 10.1016/j.theochem.2006.08.040
Theoretical Study on the Structures and Magnetic Properties of Metal String Complexes [Ni3(L)4(NCS)2] (L = dpa–, mpta–, mdpa–, mppa–)
CHEN Rong1ZHOU Wo-Hua1WU Zi-Wen1XU Xuan1,2,3,*XU Zhi-Guang1
(1School of Chemistry & Environment, South China Normal University, Guangzhou 510006, P. R. China;2Key Laboratory of Theoretical Chemistry of Environment, Ministry of Education, Guangzhou 510006, P. R. China;3Key Laboratory of Materials for Energy Conversion and Storage of Guangzhou, Guangzhou 510006, P. R. China)
Density functional theory at the BP86 level and natural bond orbital theory were used to investigate the influence of bridging ligands on the Ni―Ni interactions and magnetic coupling properties of metal string complexes [Ni3(L)4(NCS)2] (L = 1: dpa–(dipyridylamine), 2: mpta–(4-methylpyridyl-thiazolylamine), 3: mdpa–(4-methyl-dipyridylamine), 4: mppa–(4-methylpyridyl-3H-pyrrolylamine)) with potential applications in molecular wires. The following conclusions can be drawn. (1) The ground states of the complexes are antiferromagnetic (AF) singlet states, which correspond to the quintet state (HS). The energy and structure of HS is similar to AF. There are three-center-four-electronbondsalong thechains. (2) The Ni―Ni and Ni―N distances are unaffected by methyl substituents on the pyridine ring of dpa–ligands. However, substitution of the 3H-pyrrole ring or thiazole ring by the pyridine ring in mdpa–lengthens the N1―N2 and Ni―Ni distances but shortens the Ni2―N2 distance. These effects of the thiazole ring are weaker than those of the 3H-pyrrole ring. Therefore, the strength of the Ni―Ni interaction is 1 ≈ 3 > 2 > 4. (3) The predicted Jabvalues of 3 and 4are –103 and –88 cm–1, respectively. The AF magnetic coupling effects of the complexes increase with increasing Ni―Ni interaction strength: the stronger the Ni―Ni interaction, the greater the direct magnetic coupling in theorbitals along thechains. In addition, the stronger the Ni2―N2 interaction, the larger the indirect magnetic coupling involving the bridging ligand. The direct magnetic coupling is stronger than the indirect magnetic coupling.
Metal string complex; Bridging ligand; Density functional theory; Magnetic coupling; Molecular wire
O641
10.3866/PKU.WHXB201506031
Received: February 16, 2015; Revised: June 3, 2015; Published on Web: June 3, 2015.
*Corresponding author. Email: xuxuan@scnu.edu.cn; Tel: +86-13560091753.
The project was supported by the Natural Science Foundation of Guangdong Province, China (S2012010008763), Ministry of Education and Guangdong Province, China (2010B090400184), and Science and Technology Program of Guangzhou City, China (2011J4300063).
广东省自然科学基金(S2012010008763), 广东省教育部产学研项目(2010B090400184)和广州市科技攻关项目(2011J4300063)资助
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
食品科学技术学报(2022年6期)2022-12-15
化工与医药工程(2022年3期)2022-08-08
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
青岛大学学报(工程技术版)(2019年2期)2019-09-10
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
郑州大学学报(工学版)(2015年1期)2015-03-24
无机化学学报(2014年4期)2014-02-28
现代检验医学杂志(2014年4期)2014-02-02
中国兽药杂志(2012年4期)2012-11-06
