
飞秒时间分辨的光电子谱对苯S2态的超快动力学研究
2015-12-05 06:29:43沈环张冰
物理化学学报 2015年9期
沈 环 张 冰
(1华中农业大学理学院, 武汉 430070;2中国科学院武汉物理与数学研究所, 波谱与原子分子物理国家重点实验室, 武汉 430071)
飞秒时间分辨的光电子谱对苯S2态的超快动力学研究
沈 环1,*张 冰2
(1华中农业大学理学院, 武汉 430070;2中国科学院武汉物理与数学研究所, 波谱与原子分子物理国家重点实验室, 武汉 430071)
结合飞秒时间分辨的质谱技术与时间分辨的光电子影像技术对苯S2激发态的超快动力学进行了研究.苯分子吸收两个400 nm的光子被激发到S2态, 之后再用一个267 nm的光子对其进行探测. 获得的母体离子产率随泵浦探测时间延迟的变化曲线包含了两个不同的时间寿命组分. 第一个时间寿命组分(90 ± 1) fs被归纳为S2态到S1/S0态的内转换过程; 第二个时间寿命组分(5.0 ± 0.2) ps被归纳为S1态的衰减过程. 实验中观察到的第二个寿命组分小于早前的研究结果, 这表明了在S1态的衰减过程中还可能存在其他的过程. 从时间分辨的光电子影像提取得到的光电子能谱中发现了一个新的失活过程, 该过程被归结为激发态S1的振动态与“热”三重态T3之间的系间交叉过程.
时间分辨; 光电子能谱; 苯分子; 内转换; 系间交叉
1 Introduction
The excited-state dynamics of benzene have received a lot of attention since benzene is the prototype of aromatic molecules.1–16In the UV region, the first absorption band is locatedaround 250 nm. It consists of several separated peaks due to different vibronic transitions.17The second absorption band is located around 200 nm. It is a broad bump without distinguishable structure. The band origins of the first three singlet excited states were measured to be 4.76, 6.03, and 6.87 eV, respectively.17As well as the singlet states, the first three triplet states were also observed at 3.67, 4.59, and 5.47 eV by electron energy loss spectroscopy.18
Compared to the steady state spectroscopy,17,18the ultrafast dynamics of benzene is relatively unclear. As early as the 1970s, Collomon et al.1found that the lifetime of S1state ranged between 60 and 120 ns. Later, this lifetime was also found to be dependent on the symmetry of the respective excited vibrational state.2,3When the excess energy was above 3000 cm–1, a step-like increase of the decay rate was found and this sudden change was referred as “channel 3”.19Initially, internal conversion from S1state to S0state was proposed to be responsible for the increasing decay.4,11Lately, intersystem crossing to triplet state was also found to be important in this decay process.12,13
In addition to the S1state, S2state is another focus of the study. Radloff and coworkers14,15measured the lifetime of benzene on the S2state by time-resolved time-of-flight mass spectra and time-resolved photoelectron spectroscopy. They found two lifetime constants from the time-dependent yield of benzene ion. The dominant channel, which was responsible for > 90% of the decay, was due to the internal conversion from S2to S1/S0within ~50 fs. The minor component, which was relatively slow, was due to decay from S1to S0. Suzuki et al.16also studied the ultrafast decay of S2state. In order to learn the ultrafast decay of the internal conversion process, they pushed their time resolution to 22 fs and they found that the internal conversion process only took place within (48 ± 4) fs on the S2state.
In this paper, we present the ultrafast dynamics study of benzene on the S2state. The molecule is excited to the S2state by two 400 nm photons, and probed subsequently by photoionization with a 267 nm photon. The photoion yield is recorded as the pump-probe delay. The generated photoelectrons are also measured using photoelectron imaging. The time-dependent ion yield and the time-dependent photoelectron spectra allow us to trace the evolution of the excited-state dynamics simultaneously.
2 Experimental methods
The experimental setup used here has been described in detail elsewhere.20,21In brief, ~5% benzene is seeded in He carrier gas. The mixture is expanded through a pulsed valve which operates at 10 Hz. After passing a skimmer, which separates the source chamber from the ionization chamber, the molecular beam is interacted by the collinearly propagated pump and probe laser beams at the midway between the repeller and the extractor plates of the electrostatic lens. The generated photoions and photoelectrons are accelerated in the electric field and detected by a two-dimensional position sensitive imaging detector. In order to minimize the effect of the earth's magnetic field on the photoelectron trajectory, a double-layer μ-metal shield is installed along the axis of the time-of-flight chamber. A photomultiplier tube and a 100 MHz digital oscilloscope (TDS 2012, Tektronix) is used to acquire the time-of-flight mass spectrum of the photoions, while a charge coupled device (CCD) camera is used to collect the photoelectron image.
The laser source employed here is a regenerative amplified Ti:sapphire femtosecond laser system (coherent, Legend). The Ti:sapphire oscillator is pumped by the second harmonic of a CW Nd:YVO4 laser. A seed beam is generated and then amplified by a Nd:YLF laser pumped regenerative amplifier to generate a ~50 fs, 1 mJ pulse centered at 800 nm with a repetition rate of 1 kHz. The fundamental beam is split into two arms. One arm is frequency doubled to 400 nm with a bandwidth of 6 nm by a thin BBO (Beta BaB2O4) and it is used as the pump beam. The 267 nm probe beam is produced by sum frequency of the second harmonic and the fundamental. The probe beam is optically delayed with respect to the pump beam using a motorized linear translational stage (PI, M-126.CG1), which is controlled by a personal computer. The whole apparatus has a timeresolution of ~100 fs. The polarization of the pump and probe pulses is set to be vertical to the optical table and parallel to the face of the imaging detector. Both laser beams are focused using a 25-cm focal-length quartz lens. The typical power used was < 3 μJ for both beams. Each photoelectron image is accumulated over 40000 laser shots, and the background is removed by subtracting the signal at negative time delay. The photoelectron kinetic energy is calibrated using (2 + 1) resonance-enhanced multiphoton ionization of iodine atom.22
3 Results and discussion
3.1 Transient ion signal
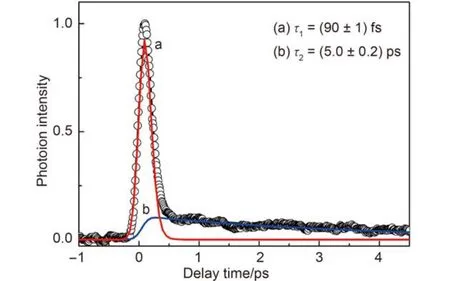
Fig.1 Yield of C6as the pump-probe time delay
Fig.1 displays the time-dependent yield of the benzene parent ion while scanning the delay between the 400 and 267 nm femtosecond pulses. Positive time delay here means that the 400 nm pulse is ahead of the 267 nm pulse. It is very obvious that the time dependent trace consists of two components, oneis a fast decay and the other one is relatively slow. After a careful data fitting, we reconstruct the trace with two exponential decay functions, i.e., a fast decay with a lifetime of (90 ± 1) fs and a slow decay with a lifetime of (5.0 ± 0.2) ps.
As shown in previous steady-state absorption measurement,17benzene did not have any absorption at 267 nm or after 300 nm. Thus, one photon excitation with either a 400 nm photon or a 267 nm photon is not going to populate the molecule to its electronic excited state. On the other hand, the ionization potential of benzene is only 9.24 eV.23Two 267 nm photons or three 400 nm photons are energetic enough to ionize the parent molecule. However, we barely find any parent ion with single pulse under our experimental condition. Therefore, we believe that exciting the molecule with two 400 nm photons followed by ionization with one 267 nm photon is the mechanism to produce the parent ion. This scheme is also supported by the excitedstate lifetime analysis and the photoelectron spectrum, as discussed in the following paragraphs.
As shown by Hiraya and Shobatake,17the energy levels of the first three excited states of benzene were located at 4.76, 6.03, and 6.87 eV, respectively. After absorbing two photons at 400 nm (hv400nm= 3.10 eV), benzene molecule is excited to the S2state. For S2state excitation, Radloff et al.14,15found two decay constants, τ1= 50 fs and τ2= 7.6 ps. The authors claimed that S2state was immediately deactivated through a two-step deactivation mechanism. A direct internal conversion to either the S1state or the vibrationally hot ground state was the fast one. The slow one was attributed to the depopulation from the vibrational excited S1state to the ground state. For the fast component, both experiments find that it decays within 100 fs. The discrepancy is due to the limited time resolution of both experiments. However, the decay of the slow component is different. Their result is 1.5 times longer than ours. This difference is important since it implies that the depopulation of the vibrational excited S1state includes other mechanisms besides the internal conversion to the ground state. Due to the similarity of the detection scheme and the probe wavelength in both experiments, it is likely that the difference is induced by the pump scheme. The vibrational-state population on the S2state and the ionization continuum in our experiment are different from theirs because we use two-photon transition instead of one-photon transition.
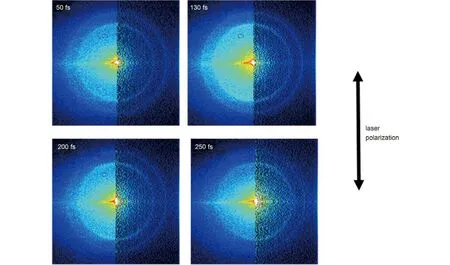
Fig.2 Transient photoelectron images in the (2 + 1') pump-probe process at different time delays
3.2 Transient photoelectron images
The photoelectron images resulted from (2 + 1') pump-probe process at different time delays are shown in Fig.2. The left part of each image is the two-dimensional (2D) raw image. These raw images are the two-dimensional projections of the three-dimensional (3D) speed and angular distributions of the photoelectrons. Giving the fact that the distributions of the photoelectrons have cylindrical symmetry around the polarization axis of the photolysis laser, a full three-dimensional photoelectron image can be reconstructed by using the basis-set expansion method (BASEX),24as shown in the right half of each image in Fig.2. Thus, the speed distributions of the photoelectrons P(υe-) are obtained from the angle integration of the reconstructed image as a function of the radial distance from the center, as shown in Fig.3. The observed images consist of two rings with differentradii. These rings are corresponding to the kinetic energy peaks that are located at 0.80 and 1.40 eV, named as peaks 1 and 2, respectively. Due to the limited S/N (signal-to-noise) ratio of photoelectron images, we neglect those peaks with photoelectron kinetic energy lower than 0.5 eV.
The kinetic energy of a photon-released electronis related to the photon energy and the corresponding internal energy of the related ion,

where, hν is the photon energy of the pump (2 × 3.10 eV) or the probe (4.65 eV),is the internal energy of the benzene ion, and the IP is the ionization potential of benzene molecule (9.24 eV).23
An energy scheme for the benzene molecule is illustrated in Fig.4. After absorbing two photons at 400 nm (6.20 eV, 0.17 eV above the band origin of the S2state), the benzene molecule is excited to the vibrational excited state of S2. After absorbing another photon at 267 nm, the maximum photoelectron kinetic energy that can be obtained by the photoelectron is 1.61 eV (2 × 3.10 + 4.65 – 9.24 = 1.61 eV). As show in Fig.3, peak 2 is centred at 1.40 eV. That means the parent ion has an internal energy of 0.21 eV. According to the Frank-Condon principle, molecule prefers to keep its nuclear geometry during the ionization process. Therefore, we attribute peak 2 to direct ionization from the vibrational excited state of S2. With increasing the time delay, the intensity of both peaks is decreasing. But peak 2shows a relative faster decay than peak 1. The faster decay of peak 2 has also been observed by Radloff et al.14,15using timeresolved photoelectron spectroscopy. They concluded that this process was the ultrafast internal conversion from S2to S1/S0state. Although the decay ratio of S2to S1is small, this ultrafast process can be observed by the time-dependent photoelectron spectroscopy.

Fig.3 Distributions of the photoelectron kinetic energy obtained from photoelectron images at different time delays

Fig.4 Schematic diagram of the relaxation dynamics for benzene with two photon excitation at 400 nm
Besides peak 2, the other peak centred at 0.80 eV (peak 1) is newly observed in our current study. As already mentioned previously, peak 1 has a different decay tendency compared to peak 2, indicating that it is not derived from S2state. The excitation of the benzene molecule with a total photon energy of 6.20 eV leads to a vibrational energy of 1.44 eV in the S1state after internal conversion from the S2state. If peak 1 resulted from this hot S1state, the photoelectron kinetic energy should be around at 0.17 eV, which has been observed by Radloff et al.14,15Therefore, we exclude the probability that the peak 1comes from the hot S1state. On the other hand, the depopulation of the S2state with a time constant of (90 ± 1) fs is due to the internal conversion to the S1/S0state. But the hot S0state cannot be observed with our probe wavelength under the current experimental condition. Thus, we also excluded the probability that peak 1 comes from the hot S0state.
Previously, the first three triplet states, T1, T2, and T3, have been observed at 3.67, 4.59, and 5.47 eV, respectively.18It is worth noticing that the ion corresponding to peak 1 has an internal energy of about 0.80 eV, which is almost equal to the interval between the total pump energy and the energy level of the T3state (2 × 3.10 – 5.47 = 0.73 eV). Thus, it is likely that peak 1 comes from ionization of the vibrational excited T3state.
As mentioned in the last section, the long-lived component with a lifetime of (5.0 ± 0.2) ps displays a faster decay than that observed by Radloff et al. That means the depopulation of the hot S1state is not only caused by the internal conversion from S1to S0state as observed previously,11,13–15but also by other ultrafast processes which may promote the decay rate. The investigations of excited benzene have shown that the lifetime of S1state depends strongly on the vibronic excess energy, regardless of whether the energy was obtained by direct or indirect excitation. When molecules were excited to 3000 cm–1above the S1state, the decay rate had a sudden increase, which may be caused by a fast intramolecular vibrational redistribution or an intersystem crossing from the optical bright state to the dark state.1–5Recently, Parker et al.13,25studied the ultrafast dynamics of excited state S1of benzene using time-resolved photoelectron spectroscopy at the onset of “channel 3”. After exciting thebenzene molecule by one 243 nm photon, an ultrafast intersystem crossing from the initially populated S1state to the optical dark triplet states T1and T2has been observed. A fast component with a lifetime of 230 fs is assigned to the quick decay from the Franck-Condon region of S1state to those triplet states. In this work, the internal conversion from the initially excited S2state to the hot S1state leads to an excess energy of 1.45 eV above the S1state. With higher excess energy, the probability of intersystem crossing between the vibrational excited S1state and those triplet states is largely increased. Since the lifetime of the long-lived component is shorter than Radloff's results, we propose that this intersystem crossing process increases the decay rate of the vibrational excited S1state. Furthermore, the peak position of peak 1 agrees well with ionization of the hot triplet state T3. Therefore, we believe that an intersystem crossing from the vibrational excited S1state to the hot triplet state T3occurred in the depopulated process.
4 Conclusions
The ultrafast dynamics of benzene on the excited state S2have been studied by femto-second time-resolved photoelectron imaging. The benzene molecule is excited to the S2state by two 400 nm photons, and probed subsequently by photoionization with 267 nm photon. The time-dependent yield of benzene ion consists of two components. The fast component is assigned to the ultrafast internal conversion from S2state to the vibrational excited S1state and the hot ground state, while the slow component is attributed to the depopulation of S1state after conversion from S2state. Two photoelectron kinetic energy peaks are extracted from the photoelectron imaging. The higher kinetic energy peak with a relative faster decay is the ionization on the S2state, while the lower photoelectron kinetic energy with a relative slow decay is attributed to the ionization from the triplet state T3after an intersystem crossing from the vibrational excited S1state. The faster decay of the S1state is rationalized by the intersystem crossing of the vibrational excited S1state to the triplet state T3.
(1)Callomon, J. H.; Dunn, T. M.; Mills, I. M. Philos. Trans. R. Soc. London A 1966, 259, 499. doi: 10.1098/rsta.1966.0023
(2)Spears, G. K.; Rice, S. J. Chem. Phys. 1971, 55, 5561. doi: 10.1063/1.1675724
(3)Wunsch, L.; Neusser, H. J.; Schlag, E. W. Chem. Phys. Lett. 1975, 32, 210. doi: 10.1016/0009-2614(75)85105-0
(4)Palmer, I. J.; Ragazos, I. N.; Bernardi, F.; Olivucci, M.; Robb, M. A. J. Am. Chem. Soc. 1993, 115, 673. doi: 10.1021/ja00055a042
(5)Sauer, P.; Xie, J. R.; Dou, Y. J. Mod. Optic. 2006, 53, 2099. doi: 10.1080/09500340600917674
(6)Worth, G. A.; Carley, R. E.; Fielding, H. H. Chem. Phys. 2007, 338, 220. doi: 10.1016/j.chemphys.2007.03.005
(7)Kuzmin, S. L.; Wesolowski, M. J.; Duley, W. W. Applied Optics 2013, 52, 8169. doi: 10.1364/AO.52.008169
(8)Qiu, X. J.; Qin, C. C.; W, J.; Zhang, B. Phys. Rev. A 2012, 86, 032505.
(9)Yin, S. H.; Liu, H. P.; Zhang, J. Y.; Jiang, B.; Wang, L.; Sha, G. H.; Lou, N. Q. Chinese Journal of Chemical Physics 2003, 16, 171.
(10)Yuan, L. W.; Zhu, J. Y.; Wang, Y. Q.; Wang, L.; Bai, J. L.; He, G. Z. Chem. Phys. Lett. 2005, 410, 352. doi: 10.1016/j.cplett. 2005.05.103
(11)Clara, M.; Hellerer, T.; Neusser, H. J. Applied Physics B-Lasers and Optics 2000, 71, 431. doi: 10.1007/s003400000347
(12)Cogan, S.; Haas, Y.; Zilberg, S. J. Photochem. Photobiol. A 2007, 190, 200. doi: 10.1016/j.jphotochem.2007.02.005
(13)Minns, R. S.; Parker, D. S. N.; Penfold, T. J.; Worth, G. A.; Fielding, H. H. Phys. Chem. Chem. Phys. 2010, 12, 15607.
(14)Radloff, W.; Freudenberg, T.; Ritze, H.; Stert, V.; Noack, F.; Hertel, I. V. Chem. Phys. Lett. 1996, 261, 301. doi: 10.1016/0009-2614(96)00972-4
(15)Radloff, W.; Stert, V.; Freudenberg, T.; Hertel, I. V.; Jouvet, C.; Dedonder-Lardeux, C.; Solgadi, D. Chem. Phys. Lett. 1997, 281, 20. doi: 10.1016/S0009-2614(97)01142-1
(16)Suzuki, Y. I.; Horio, T.; Fuji, T.; Suzuki, T. J. Chem. Phys. 2011, 134, 184313_1. doi: 10.1063/1.3586809
(17)Hiraya, A.; Shobatake, K. J. Chem. Phys. 1991, 94, 7700. doi: 10.1063/1.460155
(18)Swiderek, P.; Michaud, M.; Sanche, L. J. Chem. Phys. 1996, 105, 6724. doi: 10.1063/1.471852
(19)Penfold, T. J.; Spesyvtsev, R.; Kirkby, O. M. J. Chem. Phys. 2012, 137, 204310. doi: 10.1063/1.4767054
(20)Eppink, A. T. J. B.; Parker, D. H. Rev. Sci. Instrum. 1997, 68, 3477. doi: 10.1063/1.1148310
(21)Hua, L. Q.; Shen, H.; Hu, C. J.; Zhang, B. J. Chem. Phys. 2008, 129, 244308. doi: 10.1063/1.3047756
(22)Shen, H.; Hua, L.; Hu, C.; Zhang, B. Journal of Molecular Spectroscopy 2009, 257, 200. doi: 10.1016/j.jms.2009.08.003
(23)Nemeth, G. I.; Selzle, H. L.; Schlag, E. W. Chem. Phys. Lett. 1993, 215, 151. doi: 10.1016/0009-2614(93)89279-Q
(24)Dribinski, V.; Ossadtchi, A.; Mandelshtam, V. A.; Reisler, H. Rev. Sci. Instrum. 2002, 73, 2634. doi: 10.1063/1.1482156
(25)Parker, D. S. N.; Minns, R. S.; Phefold, T. J.; Worth, G. A.; Fielding, H. H. Chem. Phys. Lett. 2009, 469, 43. doi: 10.1016/ j.cplett.2008.12.069
Ultrafast Dynamics of Benzene on the S2State Investigated by Femtosecond Time-Resolved Photoelectron Spectroscopy
SHEN Huan1,*ZHANG Bing2
(1College of Science, Huazhong Agricultural University, Wuhan 430070, P. R. China;2State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences, Wuhan 430071, P. R. China)
The ultrafast dynamics of benzene on the S2state have been investigated by femtosecond time-resolved mass spectroscopy coupled with photoelectron imaging. The benzene molecule was excited to the S2state by two 400 nm photons, and subsequently probed by one 267 nm photon. The timedependent ion yield of the parent ion consists of two components with different lifetimes. The first component at (90 ± 1) fs is because of internal conversion from the S2state to the S1/S0state. The second one, i.e., (5.0 ± 0.2) ps, is due to decay of the S1state. The observed lifetime of the second component is shorter than previous results, indicating the existence of an additional decay process. With photoelectron spectra extracted from the time-resolved photoelectron imaging, this newly found deactivated process is assigned to intersystem crossing from the vibrational excited S1state to the hot triplet state T3.
Time-resolved; Photoelectron spectroscopy; Benzene molecule; Internal conversion; Intersystem crossing
O644
10.3866/PKU.WHXB201507061
Received: April 10, 2015; Revised: July 2, 2015; Published on Web: July 6, 2015.
*Corresponding author. Email: shenhuan@mail.hzau.edu.cn; Tel: +86-27-87282197.
The project was supported by the National Natural Science Foundation of China (21403080).
国家自然科学基金(21403080)资助项目
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
材料研究与应用(2022年5期)2022-11-07 05:36:24
煤气与热力(2021年12期)2022-01-19 05:19:30
人人健康(2021年16期)2021-12-01 07:08:33
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:24
制造技术与机床(2019年8期)2019-09-03 01:14:18
华东师范大学学报(自然科学版)(2019年3期)2019-06-24 05:29:09
中成药(2018年8期)2018-08-29 01:28:26
中成药(2018年2期)2018-05-09 07:20:09
系统工程与电子技术(2016年2期)2016-04-16 05:16:52
原子与分子物理学报(2015年3期)2015-11-24 12:49:32
