
MFI/BEA双沸石复合物的合成及其在甲烷选择性还原NOx中的应用
2015-12-05 10:25:08李彪张鸿雁郑家军秦波潘梦陈佳琪于峰王广帅李瑞丰
无机化学学报 2015年8期
李彪 张鸿雁 郑家军*, 秦波 潘梦 陈佳琪 于峰*, 王广帅 李瑞丰
(1太原理工大学能源化工与催化研究中心,太原030024) (2中石化抚顺石油化工研究院,抚顺113001)
MFI/BEA双沸石复合物的合成及其在甲烷选择性还原NOx中的应用
李彪1张鸿雁1郑家军*,1秦波2潘梦1陈佳琪1于峰*,1王广帅1李瑞丰1
(1太原理工大学能源化工与催化研究中心,太原030024) (2中石化抚顺石油化工研究院,抚顺113001)
以合成的β沸石固液混合物作为ZSM-5沸石的部分原料制备了含有ZSM-5和β沸石的双相沸石复合物MFI/BEA。采用XRD,FTIR,吡啶红外,NH3-TPD,TEM,SEM和氮吸附-脱附等对合成的材料进行了表征。结果表明后合成的ZSM-5沸石在β沸石内部孕育生长,MFI/BEA双沸石复合物中两相沸石的组成比例可以通过控制第二步晶化时间来进行有效调控;与单一的Co-ZSM-5或Co-β相比,通过离子交换法制备的Co-基复合催化剂Co-MFI/BEA在富氧条件下甲烷选择催化还原NO反应中具有高活性和高的稳定性,并且表现出较好的抗SO2毒化性能和良好的可逆性。
ZSM-5;沸石复合物;逆向生长;脱硝
0引言
将两种分子筛结合在一起形成复合分子筛[1-8],表现出了良好的协同作用和优良的催化性能[9-15]。例如,牛国兴等[13]利用正硅酸乙酯的水解,在沸石外表面形成一层无定型的二氧化硅,在模板剂的引导下促使无定型二氧化硅转化为介孔材料,并得到了以沸石为核,以介孔二氧化硅为壳的复合分子筛USY@Al-SBA-15,该材料负载NiW后在正十六烷的裂解中表现出了较单一的USY或对应的机械混合物更高的C5~C15的液体收率,且在重油的加氢裂解中表现出较对应的机械混合物催化剂更优异的催化性能;李瑞丰等[3]以工业NaY作为后合成的β沸石的铝源,制备了以Y型沸石为核,纳米多晶β沸石为壳的具有多孔结构的Y-β双相沸石复合物,与相似组成的Y与β沸石的机械混合物相比,合成出的沸石复合物在VGO(vacuum gas oil,减压馏分油)催化裂化反应过程中,表现出更高的VGO转化率和喷气燃料收率。ZSM-5沸石和β沸石在石油化工及油品精炼等方面均被广泛用作固体酸催化剂,是非常重要的催化材料。若能将ZSM-5和β沸石有机结合在一起,形成沸石复合物,则有可能获得一种新型的潜在优良催化剂。本工作采用两步晶化法,以预先晶化β沸石的固液混合物作为后合成的ZSM-5沸石的部分原料制备了一系列MFI/BEA双沸石复合物,详细讨论了沸石复合物形成的影响因素,复合物形成机制以及合成的材料的结构特点,通过离子交换法制备的Co-基复合催化剂在富氧条件下甲烷选择催化还原NO反应来评价所制备的复合沸石的催化性能。
1实验部分
1.1 试剂与仪器
四乙基溴化铵,分析纯,北京福兴化学试剂厂,wTEABr=96%;氨水,分析纯,太原化学试剂厂,wNH3·H2O=27%;铝酸钠,分析纯,上海国药试剂集团,wAl2O3= 41%,wNa2O=35%;氢氧化钠,分析纯,天津科耐恩化学试剂开发中心,wNaOH=96%;硅溶胶,工业品,青岛海洋试剂厂,wSiO2=40%;乙二胺(EDA),化学纯,天津市科密欧化学试剂开发中心;蒸馏水,自制。
日本理学Rigaku D/max-2500型X射线衍射仪(Cu Kα靶,Ni滤波,λ=0.154 18 nm),电压40 kV,电流80 mA,扫描范围为5°~35°;日本HITACHIS4800扫描电镜和JEM-200CX透射电镜观察材料的形貌和结构;采用日本岛津公司FT-IR 8400型红外光谱仪分析合成材料的骨架特征和沸石的酸性能(吡啶红外光谱);氮吸附-脱附分析采用美国康塔(Quantachrome)公司Quantachrome Quadrasorb SI型物理吸附仪进行测定;NH3-TPD测试在天津先权工贸公司的TP-6076动态吸附仪上进行。
1.2 实验过程
β沸石的合成参见文献[15],在上述合成的β沸石固液混合物中补加6 mL硅溶胶,5 mL乙二胺,调节合成体系的[OH-]在0.29~1.5 mol·L-1之间,混合搅拌成均匀白色物后装入150 mL不锈钢反应釜中,于180℃下晶化18,24,48,72 h取出,洗涤到溶液呈中性后烘干,分别命名为MFI/BEA(1),MFI/ BEA(2),MFI/BEA(3)和MFI/BEA(4);以上述晶化13 d的β沸石和采用一步法得到的ZSM-5沸石作为参照物,分别记为β和ZSM-5。
Co-基催化剂的制备:Na型分子筛首先用0.5 mol·L-1的NH4NO3溶液交换3次,m固∶m液=1∶10,110℃下干燥8 h,550℃焙烧3 h后得H型分子筛。H型分子筛用乙酸钴溶液(0.01 mol·L-1)以m固∶m液=1∶20交换,交换过程中pH值控制在5.5~6.5之间,所得固体样品经清洗,过滤,110℃干燥12 h,在空气气流中以2℃·min-1的升温速率由室温升温至550℃并保持3 h得催化剂样品。所有样品经压片,粉碎,筛分成40~60目(250~420 μm)催化剂样品备用。
催化评价:300 mg催化剂装填在内径为6 mm的石英管反应器内,He气气流中以1℃·min-1从室温升温至300℃并保持1 h对催化剂进行预处理,通入组成为2 180 μL·L-1NO+2 050 μL·L-1CH4+ 2%O2,氦气为平衡气的混合反应气。混合气流量75 mL·min-1,出口气体用装有热导检测器(TCD)的GC9A气相色谱仪在线分析,Porapak Q(不锈钢,φ 3×3 m)和5A分子筛填充柱(不锈钢,φ 5×3 m)分别用于分析N2O,CO2和N2,CO,O2,CH4,以生成N2量评价催化剂活性NO conversion=2cN2/cNO×100%,其中cN2和cNO分别为出口N2浓度和进口NO浓度)。
2结果与讨论
2.1 合成与表征
β沸石的解聚对沸石复合物MFI/BEA的形成起着至关重要的作用,在碱性体系中β沸石骨架的不稳定性能为第二步合成过程中ZSM-5沸石的生长提供充足的硅、铝物种[11,14-15]。图1表明随二次晶化时间延长,产物中ZSM-5沸石相的特征衍射峰逐渐升高,而β沸石相的特征衍射峰逐渐降低,表明MFI/BEA沸石复合物的形成是一个由BEA向MFI转变的过程[14-16]。当二次晶化时间低于18 h时,产物中还很难观察到ZSM-5沸石的特征衍射峰,这是因为在较短的时间内β沸石的解聚还不足以为ZSM-5沸石的生长提供充足的硅铝物种;当二次晶化时间为18 h时,产物中开始出现ZSM-5沸石相的特征衍射峰;当二次晶化时间达到72 h时,β沸石相的特征衍射峰几乎消失。图1还表明当二次晶化时间为18~48 h可以得到不同MFI/BEA比例的双沸石复合物。上述结果表明可以通过对二次晶化时间的控制来调变沸石复合物中两相沸石的组成比例。

图1 第二步晶化时间对复合物形成的影响Fig.1 Effect of the second crystalline time on the growth of zeolite composite
图2为MFI/BEA沸石复合物不同晶化时间的扫描电镜照片。单一的β沸石(图2A)为大小约为1 μm的多晶微球[16],ZSM-5沸石(图2F)为20~30 μm的块状晶体[15],MFI/BEA沸石复合物的晶体形貌明显不同于β沸石的球状晶体,其外表极为粗糙,整体形状趋向于棱角分明。随着晶化时间延长,产物形貌由β沸石球状晶体逐渐趋向于ZSM-5沸石的块状晶体,明显存在β沸石向ZSM-5沸石转变的痕迹。当二次晶化时间为18 h时,尽管XRD(图1)表明产物中已经出现ZSM-5沸石相的特征衍射峰,但是在SEM中还很难观察到具有ZSM-5沸石特征的粒子存在(图2B);当二次晶化时间为24 h时,图1表明ZSM-5沸石的特征衍射峰较强,但是产物中仅能见到ZSM-5沸石局部晶面(图2C),表明大多数ZSM-5沸石晶体仍被包埋在β沸石中;当二次晶化时间为48 h时,尽管能通过SEM观察到较多ZSM-5,但是晶体的大部分仍然被包裹在β沸石的纳米颗粒中(图2D)。上述结果充分表明随着晶化时间延长产物形貌逐渐由球状晶体向椭球状,并进一步向棱角分明的类似ZSM-5的块状晶体转变过程,应该归结于β沸石内部孕育的ZSM-5沸石晶体生长的结果;当二次晶化时间达到72 h,SEM观察到的主要是块状ZSM-5沸石晶体。

图2 样品的扫描电镜图Fig.2 SEM images of the samples
本课题组先前研究结果[3-4,11,14]表明,在两步晶化法制备的沸石复合物中,后合成的沸石相通常在先合成沸石外表面镶嵌生长。前面研究结果表明ZSM-5的生长可能起源于β沸石内部,存在从β沸石的内向外进行逆向生长的过程。进一步的TEM和SEM研究(图3)为β沸石内部孕育生长ZSM-5沸石提供了直接证据。在沸石复合物制备的初始阶段,发现存在很多β沸石空心球,尽管看不见明显ZSM-5沸石晶体存在,但是,此时的β沸石微球在形貌上已经由球状向块状进行转变,可能是ZSM-5这种块状晶体在β沸石内部孕育导致球状壳发生了形变(图3(1)和(2)),而此时的样品的TEM分析还很难观察到具有ZSM-5沸石特征的颗粒;MFI/BEA (2)的SEM观察中仅能观察到少量的块状颗粒,且为50 nm的纳米棒束(图3(3)和(4))。而图1的XRD揭示样品中具有很强的ZSM-5特征衍射峰,表明大多数ZSM-5沸石颗粒仍然存在β沸石内部,进一步的TEM分析(图3(9))表明,在纳米聚集体中存在约数百纳米的方块状颗粒晶体,具有ZSM-5沸石晶体典型特征,为ZSM-5沸石晶体在β沸石晶体内部生长提供了直接证据,进一步证明了ZSM-5在β沸石内部孕育生长的现象。图3(5)和(6)可以看见较多的ZSM-5沸石晶体仍然包裹在β沸石内部,仅有部分ZSM-5撑破β沸石球的包裹凸显出来,图3(10)的TEM分析进一步表明约1微米左右的多晶ZSM-5在β沸石内部孕育生长的现象。当晶化时间进一步延长到72 h,样品MFI/BEA(4)中存在大量仍被β半包裹的ZSM-5沸石晶体,见图3(7)和(8)。

图3 沸石复合物MFI/BEA不同生长阶段的SEM和TEM图Fig.3 SEM and TEM images of the different growth stages of MFI/BEA

示意图1沸石复合物ZSM-5/β形成示意图Scheme 1Schematic representation of the process for the formation of MFI/BEA
依据上述结果ZSM-5在β沸石内部孕育并逆向生长机制可以用示意图1表示。首先,β沸石受合成液中碱的腐蚀导致中空球的形成[15,17](Scheme 1 (a)→(b))。铝浓度梯度强烈影响碱性环境中沸石硅物种抽出位置和抽出量[17]。晶体内部富含硅物种,而外表面富含铝物种的特性使得在碱性环境下硅物种抽出偏爱β沸石晶体内部,最终形成空心球[18]。β沸石纳米聚集体的特性,便于第二步补加的硅物种及乙二胺等通过扩散方式进入中空的β沸石内部,并与从β沸石上解聚下来的游离硅一道,与内部创口处新形成的裸露的铝物种相互作用[19],并形成了适宜于ZSM-5沸石成核条件Scheme 1(b)→(c);ZSM-5沸石在β沸石内部生长,随着β沸石进一步解聚空心化,不断为ZSM-5生长提供养料,并促进ZSM-5不断生长Scheme 1(c)→(d);当ZSM-5晶体长大到一定程度,会撑破越来越薄的β沸石壳层Scheme 1 (d)→(e)。与传统方法合成出的纯相ZSM-5相比,沸石复合物MFI/BEA中ZSM-5沸石相晶粒要小得多,归因于ZSM-5在β沸石壳内部生长时受有限空间局域限制的作用[20-21]。上述情况说明,沸石复合物的形成是β沸石向ZSM-5沸石转化的一个过程[16],MFI/BEA仅仅是这个转变过程中的一个中间态,通过对中间状态的控制可以有效调节合成的沸石复合物中两相沸石的组成比例。
图4表明,随着晶化时间延长,沸石复合物样品中波数为570 cm-1处归属于β沸石六元环振动峰逐渐减弱,而波数为547 cm-1处归属于ZSM-5沸石的五元环振动峰逐渐增强,进一步表明MFI/BEA沸石复合物的形成是一个BEA向MFI转变的过程[16]。尽管XRD(图1)和SEM(图2D,图3(7)和(8))表明第二步晶化时间为72 h时的样品中很难观测到球状β沸石相,但是图4骨架红外表明样品MFI/BEA(4)中仍然存在β沸石的骨架特征振动峰。1 000~1 100 cm-1处的红外骨架特征振动峰通常可以用来判定沸石骨架的硅铝比。图4表明晶化时间从18 h到72 h,合成的样品的硅铝比呈现增加的趋势[14]。
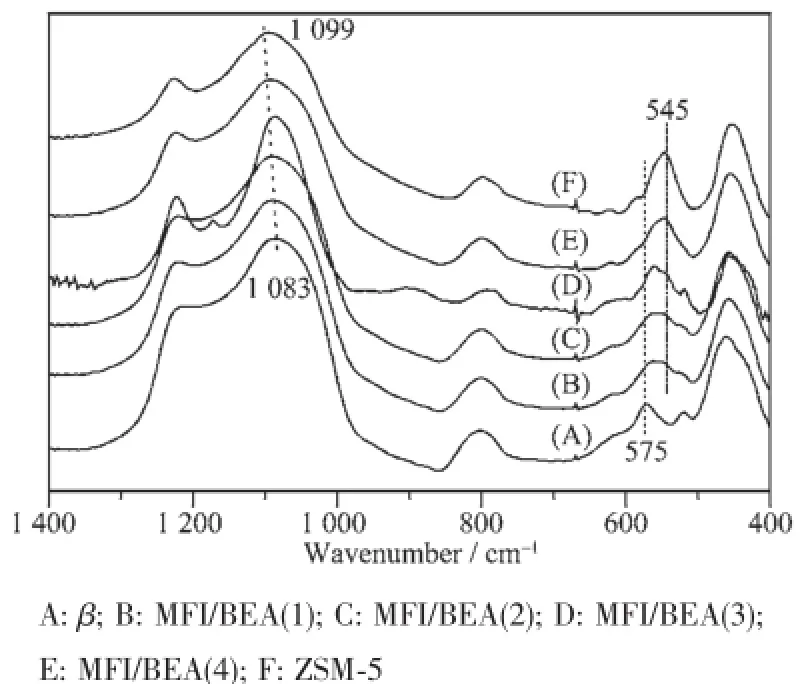
图4 样品的红外图谱Fig.4 FTIR spectra of the as-synthesized samples

表1 样品的酸性能Table1 Acid properties of the samples
吡啶红外实验结果(图5和表1)表明:与单一沸石相比,沸石复合物呈现更多的Lewis酸位。这是因为沸石复合物形成过程中伴随着β沸石的解聚,会部分破坏沸石骨架中的Si-O-Si与Si-O-Al键,导致非骨架铝物种增多,造成B酸量减少,L酸量增加[22];其次,SEM图片还表明沸石复合物中ZSM-5的晶粒远小于纯相ZSM-5,存在更多缺陷位,也造成了B酸量减少,L酸量增加现象。

图5 样品的吡啶红外图谱Fig.5 FTIR spectra of of pyridine absorbed the as-synthesized samples
图6的NH3-TPD分析表明MFI/BEA材料的弱酸中心对应的氨脱附温度介于纯β和纯ZSM-5沸石之间,而MFI/BEA沸石复合物具有比单一沸石更强的中强酸位。随着二次晶化时间延长,沸石复合物的中强酸位对应氨脱附峰值由纯β沸石的423℃移动至MFI/BEA(1)的472℃,而MFI/BEA(2)中强酸位对应氨脱附峰值则迁移至497℃,二次晶化时间进一步延长,MFI/BEA(3)中强酸位对应氨脱附峰值则下降至448℃,较接近纯ZSM-5沸石中强酸对应的氨脱附峰值。表1还表明随着晶化时间延长,沸石复合物的总酸量呈现下降的趋势,一方面归结于β沸石骨架的解聚[22];另一方面也表明第二步补加的硅物种随时间延长逐渐进入了沸石骨架,形成了更多且硅铝比组成更高的ZSM-5沸石相。

图6 催化剂的NH3-TPD曲线Fig.6 NH3-TPD profiles of the catalysts
N2吸附-脱附结果(图7)表明合成的沸石复合物为“Ⅰ型”和“Ⅳ型”等温线的结合。样品的等温线在p/p0=0.45以后存在较大的滞后环,表明存在分布较宽的介孔结构,归结于碱的作用下β沸石中硅铝物种抽出的结果[17,23],这与图2和图3的观察结果是一致的。图3(1)和(2)表明样品的β沸石壳中存在20~80 nm的中-大孔结构,为合成样品中存在明显的多孔结构[3,11,14]提供了直接证据。24 h和48 h的样品等温线在p/p0=0.8以后出现激增,应归结于沸石复合物中小的ZSM-5沸石和β沸石纳米粒子堆积形成的晶粒间介孔结构[3]。
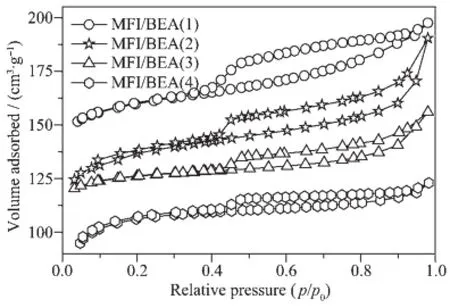
图7 样品的氮吸附-脱附等温线Fig.7 N2adsorption-desorption isotherms of the as-synthesized samples
2.2 性能评价
氮氧化物(NOx)不仅能导致酸雨和光化学雾,而且破坏大气臭氧层,直接危害人类的生存。甲烷选择催化还原NOx(CH4-SCR)是一种降低NOx排放的有效途径。单一结构分子筛如ZSM-5,Mordenite和β负载型催化剂在甲烷选择催化还原NOx反应中具有较高的催化活性。双相沸石复合物不仅能够结合两种沸石拓扑结构特点,而且在两种沸石的结合部能够形成新的金属离子交换位,有利于CH4-SCR反应进行[23]。本工作以Co-MFI/BEA催化剂考察在富氧条件下甲烷选择催化还原NO的催化活性,稳定性和抗硫性能。

图8 反应温度(T)对催化剂上NO和CH4转化率的影响Fig.8 Effect of temperature on NO conversion and CH4conversion over different catalysts
图8为Co-基催化剂上NO和CH4转化率随反应温度的变化曲线。Co-MFI/BEA催化剂上NO的转化率明显高于Co-β和Co-ZSM-5催化剂;在350℃反应明显有N2的生成,NO转化率随反应温度升高而升高,并在550~600℃达到最大值。600℃时Co-β和Co-ZSM-5催化剂上的NO转化率达到最大值,分别为40%和23%;而MFI/BEA(1),MFI/BEA(2)和MFI/BEA(3)上NO的转化率在550℃分别为58%,61.9%和54%,远高于单一沸石催化剂。
进一步提高反应温度,NO转化率逐渐下降,在650℃时,MFI/BEA(1),MFI/BEA(2)和MFI/BEA(3)上NO的转化率分别降为40%,41%和36%,但仍高于单一沸石催化剂Co-β(30%)和Co-ZSM-5(20%)。由此可见Co-MFI/BEA复合催化剂活性明显好于单一拓扑结构沸石负载型催化剂Co-β和Co-ZSM-5,且具有较宽的反应温度范围(450~650℃)。在甲烷选择催化还原NO反应中,催化剂性能与分子筛拓扑结构,酸位和活性组分所处的晶体场等有关[24-25]。与单一拓扑结构沸石β和ZSM-5相比,复合物MFI/BEA不仅能够结合这两种沸石拓扑结构的孔结构特点。图7表明复合物中成功引入多孔结构。图6表明在沸石复合物中形成了酸强度强于单一沸石的强酸位,强酸位的形成会产生一种新的Co2+离子交换位,增加了NO和NO2的吸附量,形成更多的NOx活性物种[24],可能是Co-MFI/BEA比Co-β和Co-ZSM-5催化剂具有更高催化活性的原因;表1和图6表明MFI/BEA(2)的中强酸位的量和强度是3个复合样品中最高的,可能是Co-MFI/BEA(2)具有较高催化活性的主要原因[23]。
在550℃,NO和CH4转化率在140 h内几乎保持不变,表明Co-MFI/BEA(2)催化剂具有良好的稳定性;而单一沸石催化剂Co-ZSM-5和Co-β沸石上的稳定性明显不如Co-基沸石复合物催化剂的稳定性(图9)。
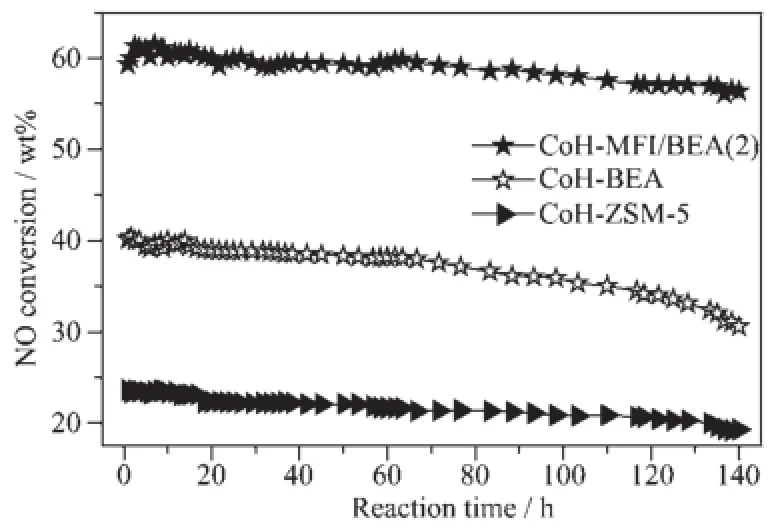
图9 Co-MFI/BEA(2)和Co-β催化剂上NO转化率(x)随反应时间(t)的变化Fig.9 Conversion of NO to N2as a function of time on stream over Co-MFI/BEA(2)and Co-β at 550℃
燃烧尾气中存在的SO2可能导致催化剂失活,因此抗SO2毒化性能是衡量催化剂性能的重要指标。SO2对Co-MFI/BEA(2)催化剂性能的影响如图10所示。在反应气中加入78 μg·g-1的SO2后,NO的转化率逐渐降低并在约3 h后稳定在50%左右。从反应气中去除SO2后,催化剂活性又较快恢复到初始水平。这些结果表明Co-MFI/BEA(2)催化剂具有一定的抗SO2毒化性能,并具有良好的可逆性能。

图10 SO2对Co-MFI/BEA(2)催化剂上NO转化率(x)的影响Fig.1 0Influence of SO2on conversion of NO to N2over Co-MFI/BEA(2)at 550℃
3结论
采用两步晶化法以先合成的β沸石作为后合成的ZSM-5沸石的硅铝源制备了沸石复合物MFI/ BEA。结果表明沸石复合物的形成是一个由β沸石相向ZSM-5沸石相转化的过程。通过控制第二步水热处理时间,可以有效控制沸石复合物中MFI和BEA两相沸石的组成比例,且能有效调节合成的沸石复合物酸性能和两相沸石的结合形态。伴随β沸石的解聚和ZSM-5在受限空间的生长,在合成的沸石复合物中引入了多孔结构。多孔结构的引入较单一沸石具有更多的路易斯酸和更强的中强酸位,导致在富氧条件下甲烷选择催化还原NO反应中Co-基复合催化剂较单一负载金属Co的沸石具有更优良的催化性能。
[1]Enterría M,Suárez-García F,Martínez-Alonso A,et al.J. Alloys Compd.,2014,583(15):60-69
[2]LI Yu-Ping(李玉萍),PAN Rui-Li(潘瑞丽),HUO Quan(霍全),et al.Chinese J.Inorg.Chem.(无机化学学报),2005,21 (10):1455-1459
[3]Zhao Q Q,Qin B,Zheng J J,et al.Chem.Eng.J.,2014,257: 262-272
[4]ZHANG Qiu(张球),TAN Wei(谭薇),ZHENG Jia-Jun(郑家军),et al.J.Inorg.Mater.(无机材料学报),2014,29(9):985 -990
[5]ZHANG Qiang(张强),XU Shao-Jun(徐少军),MENG Xiao-Jing(孟晓静),et al.Chem.J.Chinese Universities(高等学校化学学报),2013,34(4):782-787
[6]ZHANG Qiang(张强),LI Chun-Yi(李春义),SHAN Hong-Hong(山红红),et al.Chem.J.Chinese Universities(高等学校化学学报),2011,32(12):2721-2726
[7]Pirngruber G D,Laroche C,Maricar-Pichon M,et al.Microporous Mesoporous Mater.,2013,169:212-217
[8]ZHANG Lan(张兰),ZHANG Zhen-Zhong(张振中),WEI Ji-Ying(尉继英),et al.Acta Phys.-Chim.Sin.(物理化学学报), 2012,28(6):1439-1447
[9]GanjalaVSP,NeeliC K P,Pramod C V,et al.Catal.Commun., 2014,49:82-86
[10]Duan C,Zhang X,Zhou R,et al.Catal.Lett.,2011,141: 1821-1827
[11]Zheng J J,Zeng Q H,Yi Y M,et al.Catal.Today,2011,168 (1):124-132
[12]Zheng J J,Wang G S,Pan M,et al.Microporous Mesoporous Mater.,2015,206:114-120
[13]Jia L X,Sun X Y,Ye X Q,et al.Microporous Mesoporous Mater.,2013,176:16-24
[14]Zheng J J,Yi Y M,Wang W L,et al.Microporous Mesoporous Mater.,2013,171:44-52
[15]Zeng Q H,Bai X,Zheng J J,et al.Chinese Chem.Lett., 2011,22(9):1103-1106
[16]Wang D J,Liu Z N,Wang H,et al.Microporous Mesoporous Mater.,2010,132(3):428-434
[17]Groen J C,Zhu W,Brouwer S,et al.J.Am.Chem.Soc., 2007,129(2):355-360
[18]Zheng J J,Zeng Q H,Ma J H,et al.Chem.Lett.,2010,39 (4):330-331
[19]GUO Da-Lei(郭大雷),ZHENG Jia-Jun(郑家军),YI Yu-Ming (易玉明),et al.Acta Petrolei Sinica(Petroleum Processing Section)(石油学报(石油加工)),2013,29(4):591-596
[20]Bouizi Y,Rouleau L,Valtchev V P.Chem.Mater.,2006,18 (20):4959-4966
[21]Bouizi Y,Rouleau L,Valtchev V P,et al.Adv.Func.Mater., 2005,15(12):1955-1960
[22]Zheng J J,Ma J H,Wang Y et al.Catal.Lett.,2009,130: 672-678
[23]Verboekend D,Pérez-Ramírez J.Catal.Sci.Technol.,2011, 1:879-890
[24]Zhang J Q,Fan W B,Liu Y Y,et al.Appl.Catal.B:Environ., 2007,76(1/2):174-184
[25]Resini C,Montanari T,Nappi L,et al.J.Catal.,2003,214 (2):179-190
Bi-phase Zeolites Composite MFI/BEA:Synthesis and Application in Selective Catalytic Reduction of NOxby Methane
LI Biao1ZHANG Hong-Yan1ZHENG Jia-Jun*,1QIN Bo2PAN Meng1CHEN Jia-Qi1YU Feng*,1WANG Guang-Shuai1LI Rui-Feng1
(1Research Centre of Energy Chemical&Catalytic Technology,Taiyuan University of Technology,Taiyuan 030024,China)
(2Fushun Research Institute of Petroleum and Petrochemicals,SINOPEC,Fushun,Liaoning 113001,China)
Zeolite-zeolite composites composed of ZSM-5 and β zeolite crystals were synthesized by a two-step hydrothermal crystallization procedure(denoted as MFI/BEA),in which the mixture of pre-synthesized β zeolite was used as the nutrients for the growth of the post-synthesized ZSM-5 zeolite.The structural,crystalline,and textural properties of the as-synthesized materials,as well as the references ZSM-5 and β zeolite samples,were characterized by XRD,FTIR,in situ pyridine IR spectroscopy,NH3-TPD,nitrogen adsorption/desorption,TEM and SEM.The results displayed that the post-synthesized ZSM-5 zeolite unexpectedly grew within the β zeolite crystals,and the ratios of MFI/BEA in the as-synthesized zeolite-zeolite composites could be adjusted by controlling the second-step crystalline time.Catalytic performances of Co-β,Co-ZSM-5 and Co-MFI/BEA were investigated during the catalytic reduction of NO by methane in the presence of O2.As compared with the references Co-β and Co-ZSM-5,the zeolite-zeolite composites Co-MFI/BEA exhibited an excellent catalytic performance with a higher activity as well as a higher stability and an excellent sulfur-resistance.
ZSM-5;zeolite composite;reversed crystal growth;de-NOx
O643
A
1001-4861(2015)08-1563-08
10.11862/CJIC.2015.206
2015-04-22。收修改稿日期:2015-06-18。
国家自然科学基金委-中石化联合基金(No.U1463209);国家自然科学基金(No.21371129;21376157;51272169)资助项目。
*通讯联系人。E-mail:zhengjiajun@tyut.edu.cn,Tel:0351-6018384
猜你喜欢
辽宁化工(2022年8期)2022-08-27 06:02:54
云南化工(2021年10期)2021-12-21 07:33:24
煤气与热力(2021年9期)2021-11-06 05:22:56
建材发展导向(2021年13期)2021-07-28 07:14:40
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
陶瓷学报(2019年6期)2019-10-27 01:18:38
中成药(2018年7期)2018-08-04 06:04:18
中成药(2018年3期)2018-05-07 13:34:18
石油化工(2015年9期)2015-08-15 00:43:05
应用化工(2014年1期)2014-08-16 13:34:08
