
多钒氧簇化学研究进展
2015-12-01 02:36李季坤胡长文
无机化学学报 2015年9期
李季坤 胡长文
(1北京理工大学化学学院,原子分子簇教育部重点实验室,北京100081)
(2泰山学院化学化工学院,泰安271021)
多钒氧簇化学研究进展
李季坤1,2胡长文*,1
(1北京理工大学化学学院,原子分子簇教育部重点实验室,北京100081)
(2泰山学院化学化工学院,泰安271021)
多钒氧簇由于具有组成、结构和尺寸易于调控、氧化还原性以及低腐蚀性等优点,在光电、磁性、催化以及医药等领域具有广阔的应用前景,已成为多酸化学领域的研究热点之一。本文结合近年来国内外及本课题组关于多钒氧簇化学的研究,综述了多钒氧簇的合成、结构及性能研究进展,并对未来发展趋势进行了展望。
多钒氧簇;有机官能化;杂化;性质;应用
0 引言
多金属氧簇(polyoxometalates,简写为POMs)化学作为无机化学中一个重要研究领域,至今已有200年的历史,其研究对象是含有Mo、W、V、Nb、Ta等过渡元素的金属-氧簇合物(metal-oxygen clusters)[1-3]。作为多金属氧簇的一个重要分支,多钒氧簇(polyoxovanadates,简写为POVs)因其多样的结构以及在催化、磁性、光电和医药等领域潜在的应用前景成为POMs化学的研究热点之一[4-6]。POVs系列簇合物可分为两大类,即同多钒氧簇和杂多钒氧簇。同多钒氧簇骨架结构通常仅由V和O原子构成。而杂多钒氧簇是把杂原子引入到POVs骨架中,自第一例[MV13O38]7-(M=Mn,Ni)的结构被确定以来,已有大量的杂多钒氧簇化合物被相继报道,从而大大拓展了传统POVs的应用范围[7]。杂多钒氧簇相关内容请参考Hayashi[8]关于杂多和缺位多钒氧簇以及本课题组关于取代钒氧簇研究进展的综述文献[9],本文对此部分仅做简要介绍,重点综述近年来同多钒氧簇的研究进展。
POVs的结构多样性主要归因于以下两点:(i)V原子具有多变的配位构型,包括VO4四面体、VO5四方锥、VO6八面体等,它们可以单独也可以混合存在于POVs结构中。(ii)V元素主要存在3种氧化态(VⅢ、VⅣ、VⅤ),它们同样可以不同比例混合存在于POVs骨架结构中。POVs合成过程中比较重要的影响因素包括起始原料钒源、有机配体的选择、反应液的pH值及反应温度等。本文从合成方法、结构特征以及功能特性方面综述了近年来多钒氧簇化学研究进展,并对其未来的发展趋势进行了展望。
1 同多钒氧簇
同多钒氧簇种类繁多,本部分主要从三大体系分别予以讨论:(1)抗衡离子型多钒氧簇;(2)有机官能化多钒氧簇;(3)金属有机单元修饰多钒氧簇。上述3个体系中具有确定结构的同多钒氧簇按照钒核的数目以及抗衡离子和有机配体的类型分别归纳于表1~3。

表1 抗衡离子型多钒氧簇Table 1POVs clusters of counterion-type

续表1
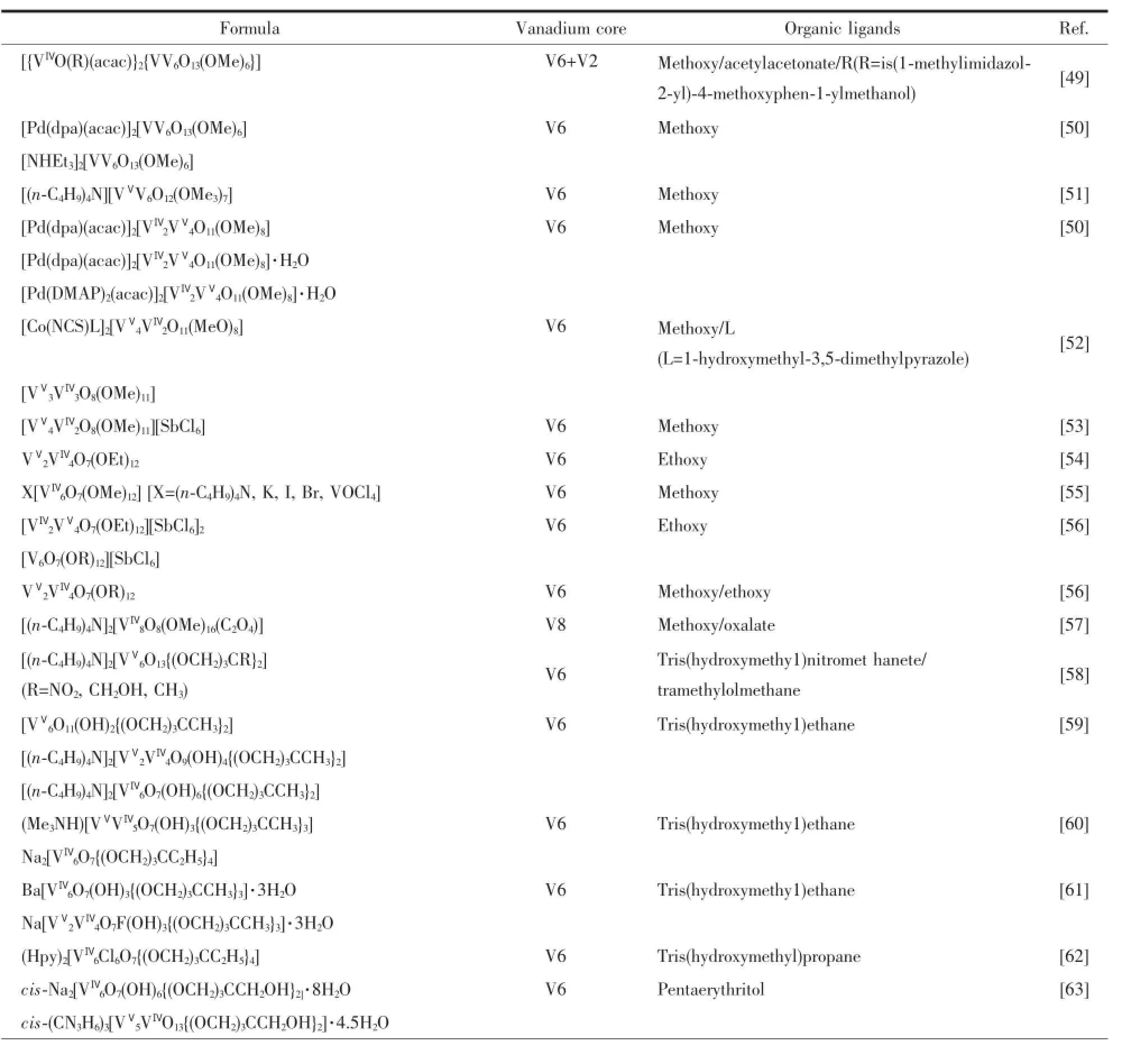
表2 有机官能化多钒氧簇化合物Table 2Organic-functionalized POVs clusters

续表2

续表2
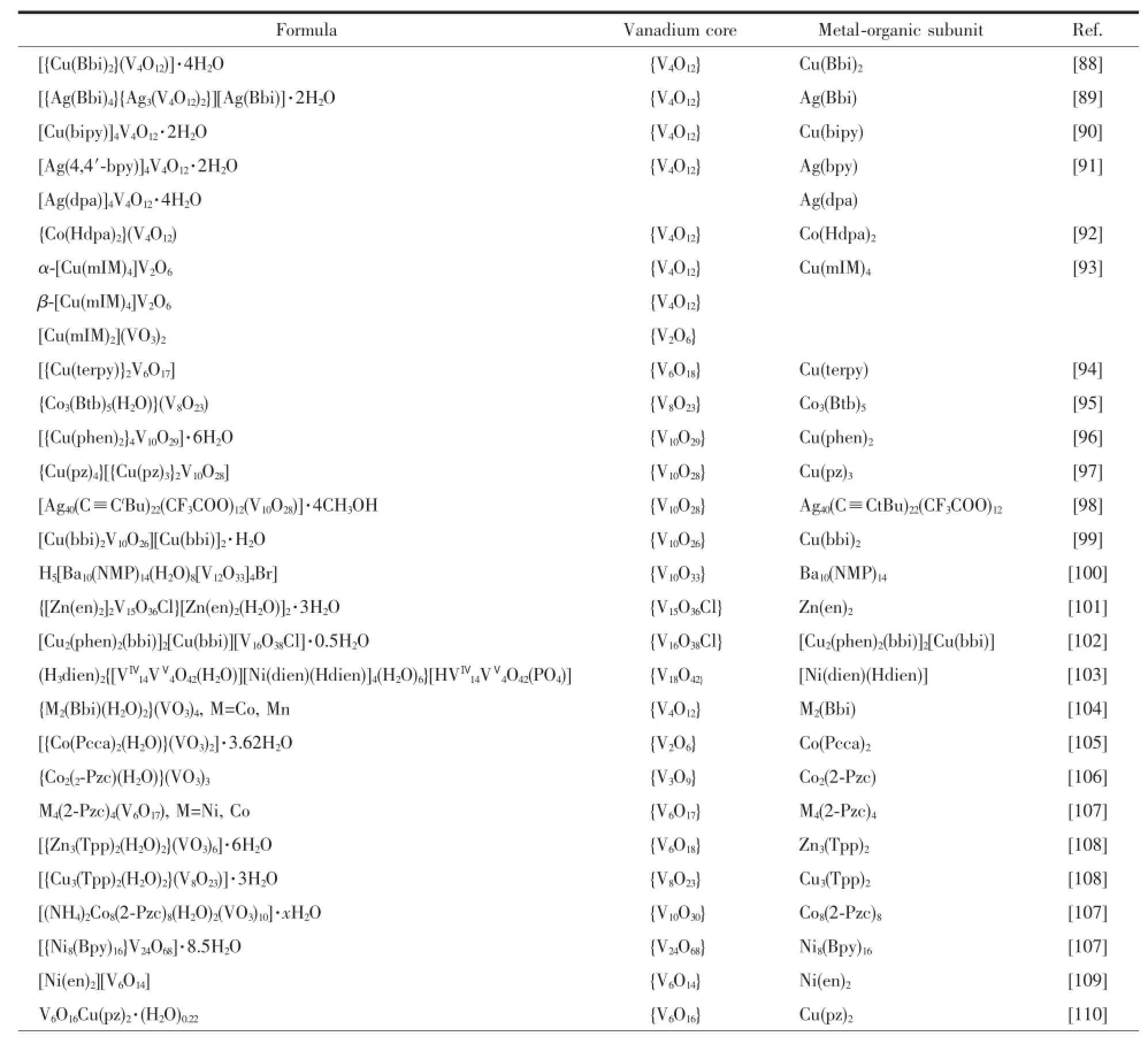
表3 金属-有机单元修饰的多钒氧簇Table 3Mtal-organic unit modified POVs clusters

续表3
1.1抗衡离子型多钒氧簇[10-46]
同多钒酸根阴离子的种类很多,迄今,最高核同多钒氧簇已达到{V34}核[32]。然而,由于大多钒氧簇阴离子的端O或桥O具有较好的配位能力,当抗衡阳离子为金属或金属-有机单元时很容易与钒氧簇配位,直接修饰在同多钒酸根阴离子上,只有金属或金属-有机单元的配位点被溶剂分子或配位能力很强的螯合类含氮配体完全占据,将金属离子包裹起来,才能使钒酸根阴离子成为孤立的抗衡离子型多钒氧簇。此类化合物一般采用常规方法在水溶液或有机溶剂中合成,典型的化合物列于表1。
1.1.1简单抗衡阳离子型多钒氧簇
简单抗衡离子型多钒氧簇是指抗衡阳离子为碱金属、过渡金属、铵(有机胺)等离子的POVs。此类型多钒氧簇的共同特点是抗衡阳离子的剩余配位点都被溶剂分子占据,无法进一步与多钒氧簇配位。目前,最高核的同多钒氧簇K10[VⅣ16VⅤ18O82]·20H2O[32]就属于这一类型。此类钒氧簇根据合成所需钒源的不同可以在水溶液或有机溶剂中合成,在水溶液中选用溶解度比较好的钒源,例如NH4VO3[10,19-20,28,31]、NaVO4[12]、NaVO3[21]、KVO3[13,18,32]、V2O5[14-16]、VOSO4[17,27,30]、CsVO3[25]以及(NH4)3[VS4][25]等,相应的钒氧簇都是通过调控水溶液的pH值合成得到。{V19}核钒氧簇(NH4)8[H9V19O50]·11H2O也可以用作水溶液中合成钒氧簇[NEt4]5[H2V18O44(N3)][29]、[NEt4]6[HV22O54(ClO4)][29]的钒源。而有机溶剂中溶解度较好的[(n-C4H9)4N]3[V10O28H3]、VO2(acac)和[Net4]2[VOCl4]等可以作为起始钒源在有机溶剂中合成种类繁多的多钒氧簇,例如[(n-C4H9)4N]3[V13O34][23]、[Et4N]5[V14O36Cl][24]、[NEt4]5[V15O36]·1.28MeCN[26]等。
1.1.2金属配合物抗衡阳离子型多钒氧簇
金属配合物抗衡阳离子型多钒氧簇是指抗衡阳离子为具有较强配位能力的有机配体修饰的金属离子的POVs。在此分别以簇状的[V4O12]4-[33,35]、[V10O28]6-[36-38]、[V15O36]5-[37,39-40]为例介绍一下该类型的POVs。
[V4O12]4-簇的表面有具有较强配位能力的8个端O,而当金属离子与一些含N及含O的螯合类配体,如1,10-phen(phenanthroline)、bipy(bipyridine)、dpa (2,2′-dipyridylamine)、en(ethylenediamine)、ox(oxalate)等配位后就能阻断与钒氧簇阴离子的配位,成为孤立的金属配合物抗衡阳离子。我们2003年合成的[Ni(phen)3]2[V4O12]·17.5H2O[34]就是基于[V4O12]4-簇的金属配合物抗衡离子型多钒氧簇的一个典型例子。在此化合物结构中,每个Ni2+分别与3个1,10-phen配体配位形成[Ni(phen)3]2+金属-有机阳离子,因此Ni2+的6个配位点均被phen配体的N原子占据,阻断了其与[V4O12]4-簇的配位,使其仅作为抗衡阳离子起到平衡电荷的作用。而相邻的[V4O12]4-阴离子簇通过与晶格H2O形成H键拓展成稳定的3D超分子网络结构。此类型化合物有[{Co(phen)2}2V4O12]、[Zn (2,2′-bipy)3]2V4O12·11H2O[41]、[{Co(3,3′-bipy)2}2V4O12][42]、[H2pn][Mn2(ox)(V4O12)][43]、[Ni(Hdpa)2V4O12][44]、[Mn(Hen)2V4O12][45]、以及[Cu3(trz2)V4O12][46]等。
2006年,辽宁师范大学由万胜课题组报道了一例含有[Co4O4]立方烷簇的[Co4O4(dpaH)4(CH3COO)2]2+为抗衡阳离子,游离的[V4O12]4-为抗衡阴离子的化合物[Co4O4(dpaH)4(CH3COO)2]2V4O12·5H2O[35]。2,2′-二吡啶胺与Co2+螯合配位,乙酸根桥连了2个Co2+离子,而晶体学独立的4个Co2+通过O原子连接成具有立方烷结构的[Co4O4]四核簇。这是首例将[Co4O4]立方烷簇与钒氧簇结合的例子,这为具有新颖结构的多钒氧簇材料的合成开辟了新的方向。
基于{V10}簇的金属配合物抗衡离子型多钒氧簇合物的阴离子一般为紧密堆积型的[V10O28]6-。2002年,东北师范大学王恩波课题组通过水热法合成了一例[Ni(phen)3]2+为抗衡阳离子的多钒氧簇化合物[Ni(phen)3]2[V10O26][37],而[VⅣ2VⅤ8O26]4-是1个球状中空型混价的同多钒酸根阴离子,一般具有笼状的阴离子内部都会填充一些体积相对较小的阴离子如Cl-、CO32-、ClO4-以及中性的分子如H2O等,而中空型的笼状同多钒氧簇还是较为少见。
基于{V15}簇的金属配合物抗衡离子型多钒氧簇合物大都含有紧密堆积型的[V15O36Cl]5-抗衡阴离子,其中的Cl-填充到阴离子簇的内部对笼形钒氧簇的组装起到导向性作用。而东北师范大学彭军课题组于2007年合成了一例同时含有[V15O36(Cl)]和[V17O40(Cl)]簇的化合物[Ni(phen)3]2{[V15O36(Cl)]0.5[V17O40(Cl)]0.5}·H2O[39]。该化合物由2个[Ni(phen)3]2+金属-有机阳离子和2个半占有的[V15O36(Cl)]和[V17O40(Cl)]簇组成。多钒酸根的平均聚合度为[V16O38(Cl)]4-。这是首例关于[V17O40(Cl)]的报道,而也是同一化合物中同时含有[V15O36(Cl)]和[V17O40(Cl)]簇的首例报道。
1.2有机官能化钒氧簇
有机配体官能化大大促进了多钒氧簇的发展。与多钼和多钨氧簇有机官能化的大量研究相比,POVs在这一领域的研究要少很多。有机官能化POVs把金属-氧簇的独特性能与具有高活性官能团的有机配体结合起来扩展了POVs的应用范围。POVs与有机配体通过共价键结合能增加其在有机溶剂中的溶解度、调控氧化还原性能、提高其稳定性,拓展其在催化、磁性、光电等功能材料方面的应用。本部分主要介绍烷氧基、有机羧酸、有机砷酸、有机膦酸、含N配体有机官能化的POVs,典型的例子列于表2。
1.2.1含氧配体有机官能化多钒氧簇
含氧有机配体主要包括醇、酚、有机羧酸、有机砷酸、有机膦酸等。研究结果表明,此类化合物大部分都是在非水溶剂中合成的且对温度比较敏感,化合物中V原子的价态多变,许多化合物中出现混合价态。合成条件不同,V的聚合度也不同,常形成“笼”、“环”、“饼”等簇状结构,也会生成更高维度的链或层状结构。早期烷氧基和有机砷酸官能化的POVs请参考1995年Khan和Zubieta的综述[47],而关于有机磷酸和有机羧酸官能化的POVs请参考Zubieta和Dolbecq的文献[47-48],本文仅介绍近10年来此类化合物的研究进展。

图1 甲氧基化Lindqvist型多钒氧簇结构Fig.1 Methoxo-substituted Lindqvist POVs
1.2.1.1醇/酚配体有机官能化多钒氧簇
烷氧基-POVs是有机官能化多钒氧簇中最大的一个分支。烷氧基-POVs的化学性能介于金属氧化物和金属烷氧化物之间。通过改变POVs中烷氧基团的数目以及VⅣ/VⅤ的比例可以实现对金属-氧簇性能和组成的调控,而这对于其在催化化学中的应用是非常重要的。而烷氧基配体可以分为单齿和多齿配体两大类。合成烷氧基-POVs的钒源可以是氧化钒、钒酸盐、有较大抗衡阳离子的多钒氧簇以及钒酸酯类化合物VO(OR)3等。
单齿烷氧基配体取代Lindqvist型多钒氧簇[V6O19]8-的桥O是补偿负电荷及稳定多阴离子簇的有效途径。利用这种策略可以合成因V5+较小的离子半径以及簇合物表面的高电荷而导致的水溶液中不能稳定存在的Lindqvist型[V6O19]8-钒氧簇。甲氧基和乙氧基由于其较小的位阻和较强的配位能力而成为取代[V6O19]8-的桥O的优选配体。如图1所示,到目前为止,[VⅤ6O13(OMe)6]2-[49-50],[VV6O12(OMe)7]-[51], [VⅣ2VⅤ4O11(OMe)8]2-[50,52],[VⅣ3VⅤ3O8(OMe)11][53]和[VⅣ2VⅤ4 O8(OMe)11]+[53],[VⅣ4VⅤ2O7(OEt)12][54]和一系列的[V6O7(OMe)12](R=-CH3,-C2H5)[56]已经被合成并确定了结构。值得一提的是,[V6O7(OMe)12]中VⅣ/VⅤ的比例可以在合成过程中通过加入氧化剂或还原剂实现调控。Hartl和Daniel′s合成了一些含有金属抗衡阳离子以及中性多钒氧簇的化合物[VⅣ(4-n)VⅤ(2+n)O7(OR)12]n+[SbCl6]n(R=-CH3,n=0,1;R=-C2H5,n=0,1,2)[55-56],[V6O8(OCH3)11][SbCl6],[VⅣ4VⅤ2O7(OR)12](R=-CH3,-C2H5)[53]。
单齿烷氧基配体取代的Lindqvist型钒氧簇[V6O19]n-的抗衡阳离子一般为质子化的有机胺或者碱金属离子。2002年,德国的Krebs课题组合成了第一例金属-有机化合物为抗衡阳离子的双支撑型POVs化合物[{VO(bmimpm)(acac)2}{V6O13(OMe)6}][49](bmimpm=bis(1-methylimidazol-2-yl)-4-Me-thoxyphen-1-ylmethanol),化合物中[V6O13(OMe)6]2-与[VO]2+的化合物通过配位键连接。2012年合成的八甲氧基取代的[Co(NCS)L]2[VⅤ4VⅣ2O11(Me3O)8][52](L=1-hydroxymethyl-3,5-dimethylpyrazole)是第一例过渡金属-有机化合物为抗衡阳离子且仅通过阴阳离子静电作用结合的Lindqvist型钒氧簇。
2015年我们合成了4个新型的贵金属Pd配合物为抗衡阳离子的Lindqvist型甲氧基化多钒氧簇化合物[50][Pd(dpa)(acac)]2[V6O13(OMe)6](a),[Pd(dpa) (acac)]2[VⅣ2VⅤ4O11(OMe)8](b),[Pd(dpa)(acac)]2[VⅣ2VⅤ4O11(OMe)8]·H2O(c),和[Pd(DMAP)2(acac)]2[VⅣ2VⅤ4O11(OMe)8]·H2O(d)(DMAP=4-dimethylaminopyridine;acac =acetylacetone anion),如图2所示,并研究了其催化性能。在所有化合物中钒氧簇阴离子仅通过静电作用与Pd配合物连接。
值得一提的是,我们可以通过控制反应条件,实现钒氧簇的调控合成,例如,通过调控三乙胺的用量我们可以分别得到六和八甲氧基取代的钒氧簇,而通过调控结晶温度,我们可以制备由甲氧基取代位置不同而导致的同分异构的钒氧簇阴离子α-[V6O11(OMe)8]2-和β-[V6O11(OMe)8]2-。而β-[V6O11(OMe)8]2-是一例前所未有的Lindqvist型多钒氧簇阴离子,如图3所示。

图2 钯配合物抗衡阳离子型Lindqvist甲氧基化多钒氧簇合物的控制合成[50]Fig.2 Controlling synthesis of Pd-complex combined methoxo-substituted Lindqvist POVs
酚羟基官能化的Lindqvist型POVs比较少见,典型的例子是2008年西班牙Luneau课题组合成的一系列有机/无机胺为抗衡阳离子的杯四芳烃官能化的POVs化合物(cat)[VⅢVⅣ5O6(OCH3)8(calix) (CH3OH)](cat=Et4N,NH4,PyH,Et3NH)[71],其Lindqvist型钒氧簇阴离子中12个桥O中有8个被μ2-OCH3取代,有4个被calix配体的4个酚羟基O取代。其中三价VⅢ原子的端氧来自于甲醇,而脱质子的杯芳烃对其起到了保护作用。这是第一例含有VⅢ/VⅣ混合价态的Lindqvist型钒氧簇化合物。

图3 化合物b和c中的同分异构POVs阴离子簇Fig.3 Isomeric POVs anion clusters in b and c
多元醇官能化的Lindqvist型POVs以(HOCH2)3CR(R=CH3,C2H5,CH2OH)类型的配体为代表。如图4所示,此类配体可以取代POVs簇上的6个[58-59,63-67]、9个[60-61]或12个桥氧[62]与V以三角形的方式配位。自1990年Zubieta课题组利用三醇类配体与[(n-C4H9)4N][H3V10O28]反应合成第一例多元醇官能化的POVs[(n-C4H9)4N]2[V6O13{(O-CH2)3CR}2](R=NO2,CH2OH, CH3)以来[58],该课题组以及Müller,Crans课题组大大扩展了这一领域的研究,合成了一系列此类型的POVs化合物,详见表2。而采用的合成策略主要包括以下三类:(1)在相同的起始反应物[(n-C4H9)4N]2[VⅤ6O13{(OCH2)3CCH3}2]的反应体系中加入还原剂1,2-二苯肼,甲基苯肼,HBF4·O(C2H5)2,生成有混合价态或者全还原价态钒的[VⅤ6-nVⅣnO13-n(OH)n{(OCH2)3CCH3}2]2-(n=2,4,6)[59]。(2)用钒的氧化物为起始原料,在矿化剂及水热反应条件下可以得到9个桥氧被取代的[V6O7(OH)3{(OCH2)3CCH3}3]n-(n=1,2)[60-61]和12个桥氧完全被取代的[V6O7{(OCH2)3CC2H5}4]2-[60]。(3)μ3-F或Cl取代{V6O19}的中心O或端O基团而合成有趣的[V6O7F(OH)3{(OCH2)3CCH3}3]-[61]或[V6Cl6O{(OCH2)3CC2H5}4]2-钒氧簇[62]。
对Lindqvist型钒氧簇的后修饰也是合成新型官能化多钒氧簇的有效途径。美国的Hill课题组利用三醇类烷氧配体取代的[V6O13{(OCH2)3C (NHCH2C6H4-4-CO2)}2]4-与Tb(Ⅱ)反应得到一个有3D网状结构的化合物Tb[V6O13{(OCH2)3C(NH2CH2C6H4-4-CO2)}{(OCH2)3C-(NHCH2-C6H4-4-CO2)}2],并研究了其在以O2为氧化剂的硫醇氧化生成二硫化物反应中的催化特性[65]。
2011年,清华大学魏永革课题组利用(Bu4N)2[V6O13{(OCH2)3CCH2OH}2]与酸酐发生酯化反应得到一系列两头有长链烷烃的POVs[67],如图5所示。同一课题组于2012年利用六核钒氧簇与具有长链的脂肪酸经酯化反应得到有机-无机杂化的多钒氧簇[V6O13{(OCH2)3CCH2OOC(CH2)16CH3}2]2-。当H质子取代抗衡阳离子TBA后,该化合物能发射蓝色荧光,这是首次在POMs化合物中发现由抗衡离子决定的发射荧光现象[64]。
1.2.1.2有机酸配体官能化多钒氧簇
参与POVs官能化的有机酸配体包括有机羧酸、有机砷酸、有机膦酸等。由于含有多个含O配齿,因此有机酸官能化的POVs表现出多样的结构特征,包括离散的“环”、“笼”等簇状或层状以及3D框架结构等。
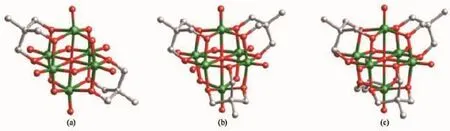
图4 (a)2、(b)3和(c)4个(HOCH2)3CCH3配体官能化的Lindqvist型POVs簇[58-67]Fig.4 (a)Two,(b)three and(c)four(HOCH2)3CCH3functionalized POVs clusters
有机羧酸官能化的POVs表现出多种多样的结构特征,其中V的价态以混价的居多。与羧基配位后,钒几乎都是VO6八面体配位模式,VO5四方锥和VO4四面体配位模式则少有报道。而一些其它的有机配体如一元醇、乙酰丙酮、有机膦酸等常协助有机羧酸构筑POVs化合物[72-74]。2007年英国的McInnes课题组报道了一系列由苯甲酸和乙醇修饰的{V11}、{V13}、{V16}和{V18}[72-73]高核钒氧簇。Hartl课题组也报道了一例由方酸和甲醇修饰的二十四核钒氧簇化合物[(n-Bu)4N]8[VⅣ24O24(C4O4)12(OMe)32][74]。该立方形结构的钒氧簇,其顶点为八个甲醇修饰的{VⅢ}簇,而十二条棱分别由12个方酸根桥连而成。
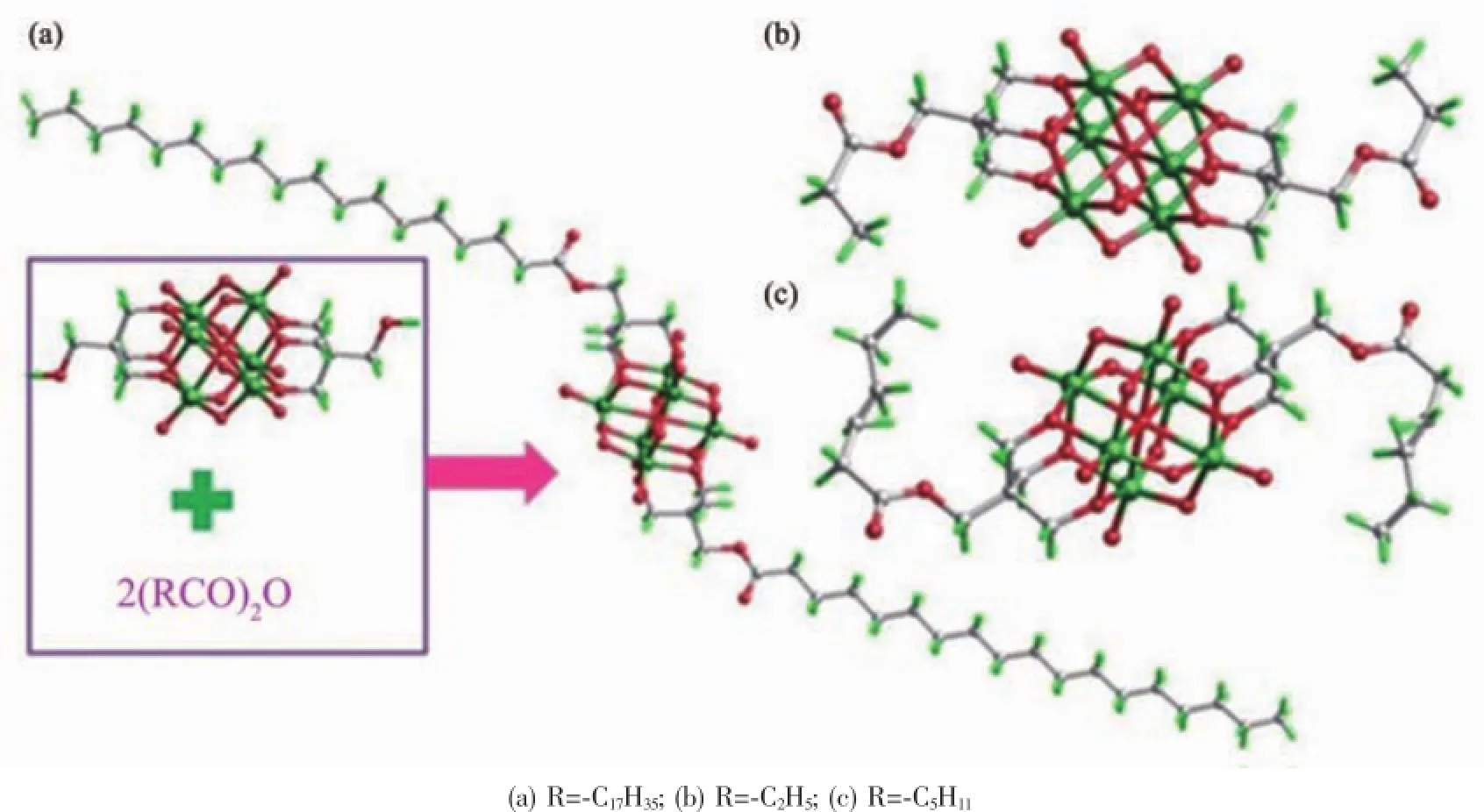
图5 不同长链烷烃修饰的Lindqvist型POVs的酯化合成及结构[67]Fig.5 Esterification synthesis and structures of Lindqvist type POVs modified by different alkanes
有机羧酸官能化的POVs以单羧基的配体为主。含2个及以上羧基的配体参与有机官能化POVs的例子很少见。2002年,法国的Riou课题组合成了1个间苯二甲酸修饰的具有V3结构片段的3D化合物[V(H2O)]3O(O2CC6H4CO2)3·(Cl,9H2O)[75]。结构中V3亚单元由3个八面体配位的VO6共顶点于1个O2-形成,而每1个VⅢ与1个μ3-O、1个H2O及4个羧基O配位,每个3核簇与6个二羧酸配体连接成3D框架结构。
2014年,美国的Zaworotko课题组合成了三例非常罕见的由均苯三羧酸与四核和五核的钒氧簇自组装形成的有机官能化纳米球POVs(NH2Me2)6[(V4O8Cl)6(BTC)8]、(NH2(Butyl)2)12[(V5O9Cl)6(BTC)8]和(NH2Me2)8[(V4O8Cl)3.8(V5O9Cl)2.2(BTC)8](BTC=1,3,5-benzentricarboxylate)[76],较大的内表面积表明这些纳米球有刚性的孔道,如图6所示。而纳米球功能化的外表面可以通过氢键或配位键进行进一步的修饰而制备立方形的网状结构化合物。此类材料预期在催化及传感器领域会有较好的应用前景。
有机砷酸官能化的POVs多数为零维的离散簇结构[77-79]。簇中V的价态多为混价,V的配位模式多为VO4四面体、VO5四方锥和少量的VO6八面体。爱尔兰的Schmitt课题组利用加还原剂及调节pH值的方法制备了具有杯状结构的[V5O9(O3AsC6H4NH2)4]5-和笼状结构的[V12O14(OH)4(H2O)2(O3As-C6H4NH2)10]4-[77]。该组在2011年又合成了具有高对称型结构的{V16As10}、{V20As8}以及{V24As8}的钒氧簇[79]。
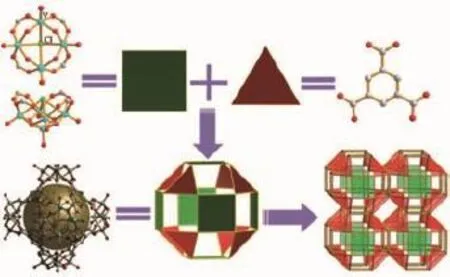
图6 均苯三酸官能化POVs纳米球的组装机理[76]Fig.6 Assembly mechanism of trimesic acid functionalized POVs with nanostructure
2012年,本课题组合成了4个有机砷酸官能化的多钒氧簇合物[H6VⅤ2VⅣ14O24(OH)8(O3AsC6H4-4-NH2)8] ·4DMF·12H2O,(Me2NH2)8·[(VO)16(OH)8O8(O3AsC6H4-4-NH2)8]·DMF·12H2O,(Me2NH2)8·[(VⅣO)16(OH)8O8(O3AsC6H4-2-NH2)8]·DMF·H2O和(Me2NH2)8·[(VⅣO)16(OH)8O8(O3AsC6H5)8]·3DMF·4.5H2O[80]。研究表明取代基及反应温度对有机砷官能化POVs的合成均有一定影响,如图7所示。
有机膦酸也是POVs有机官能化的一类重要的配体,并构筑了大量“环”、“笼”及“层”状的结构[77,81-84]。与有机单膦酸相比,有机二膦酸官能化的POVs就少很多。2008年,爱尔兰的Schmitt课题组合成了2个“胶囊”状的有机膦酸参与构筑的POVs,[H2V10O18(O3PC6H4PO3)4]8-和[H2V10O18(O3PC12H8PO3)4]8-[77]。这2个化合物的结构类似,都是4个芳香二膦酸根连接2个{VⅤO(μ3-O)4VⅣ4O12}单元。通过改变有机膦酸的长度,2个“胶囊”会展现出不同的尺寸。这种由有机膦酸构筑的尺寸可调的“胶囊”状POVs化合物在文献中是不多见的。2014年,该课题组又在此领域取得了最新进展,他们用刚性的二膦酸配体bis(4-phosphonatophenyl)ethyne和bis(4-Phosphonatophenyl)butadiyne)合成了尺寸可调控的“胶囊”状的有机官能化POVs H6Na6[(N3)2V10O18(O3PC14H8PO3)4]·42H2O和H6Na6[(N3)2V10O18(O3PC16H8PO3)4]·40H2O[84],如图8所示。预期此类化合物在催化剂分子识别领域会有比较好的应用前景。

图7 反应温度和有机砷酸配体类型对POVs结构的影响示意图[80]Fig.7 Schematic representation of influence of the reaction temperature and ligands on organoarsonato POVs
1.2.2含氮配体有机官能化多钒氧簇
在有机官能化POVs合成中,含N配体一般用作其它含O配体(醇、酚、有机酸)的辅助配体与V配位。而没有含O配体参与的N原子直接与POVs配位的例子非常少见。2005年,中国科学院福建物质结构研究所的郭国聪课题组合成了一例手性混合价态的有机胺官能化的POVs化合物[VⅤ2VⅣ3O11(dien)3](dien:diethylenetriamine),这是第一例中性的五核钒氧簇化合物[86]。
2013年,本课题组以咪唑类化合物为有机配体和溶剂合成了3个咪唑衍生物官能化的多钒氧簇VⅤ6O15(mIM)8,VⅣ2VⅤ4O14(mIM)8,VⅣ2VⅤ4O14(eIM)8(mIM= 1-Methylimidazole;eIM=1-Ethylimidazole)。化合物VⅤ6O15(mIM)8由4个甲基咪唑官能化的VO2(mIM)2通过共角连接的二聚体V2O7拓展为V6簇。通过改变投料中的钒源化合物及咪唑衍生物配体的类型而得到化合物VⅣ2VⅤ4O14(mIM)8和VⅣ2VⅤ4O14(eIM)8,这2个化合物都含有混合价态的V6簇。有趣的是,化合物VⅣ2VⅤ4O14(mIM)8和VⅣ2VⅤ4O14(eIM)8可以在特定条件下实现单晶-单晶的转化[87],如图9所示。
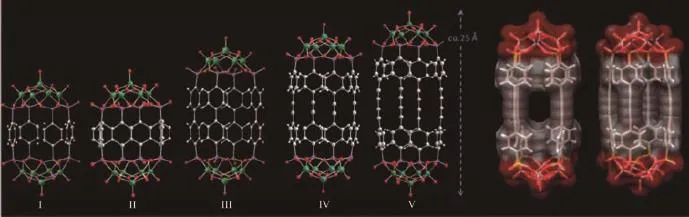
图8 有机二膦酸配体官能化的不同尺寸“胶囊”状的POVs[77,84]Fig.8 Organic diphosphonates ligands functionalized POVs with“capsule”shape and different sizes
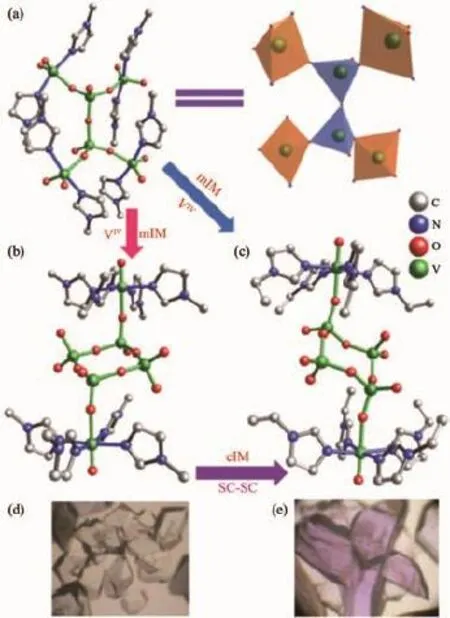
图9 化合物之间的相互转化及光学显微镜照片[87]Fig.9 Molecular structures of compounds VⅤ6O15(mIM)8(a),VⅣ2VⅤ4O14(mIM)8(b),VⅣ2VⅤ4O14(eIM)8(c)and the phase transformations among them;Optical micrographs of VⅣ2VⅤ4O14(mIM)8(d), VⅣ2VⅤ4O14(eIM)8(e)
1.3金属-有机单元修饰的多钒氧簇
由于多钒氧簇表面大量的端O和桥O具有不同程度的配位能力,且其一般带有数量不等的负电荷,因此POVs阴离子簇很容易结合带正电荷金属离子,而金属离子剩余的配位点又可以进一步与有机配体配位,这样就可以把有机配体和过渡金属同时引入到POVs中形成具有簇状[88-103]、链状[104-108]和层状[109-113]同多钒-氧亚单元结构的钒氧簇化合物,典型的化合物列于表3。此类化合物以钒氧簇聚合度的不同分为[V2O6]4-[93,105]、[V4O12]4-[88-93,104,111-113]、[V6O18]6-[94,108]、[V6O17]4-[107]、[V6O16]2-[110]、[V6O14]2-[109]、[V8O23]6-[95,108]、[V10O29]8-[96]、[V10O26]4-[99]、[V10O28]6-[97-98]、[V15O36]5-[101]、[V16O38]4-[102]、[V18O42]5-[103]等。此类化合物详细合成及结构描述请参考2014年的综述文献[154]。本文结合我们课题组最近的工作对近几年金属有机单元修饰多钒氧簇的典型例子作简要介绍。
2012年,德国Ktreb课题组合成了一例58核的杂金属钒氧簇合物H5[Ba10(NMP)14(H2O)8[V12O33]4Br][114](NMP=N-methyl-2-pyrrolidone)。以Br-为中心,周围是10个BaO6八面体共边共角连接形成十核Ba簇,在十核Ba簇的周围又通过Ba-O-V连接了4个前所未有的紧密堆积型[V12O33]6-,最终形成Ba10V48杂金属簇,而簇的外围通过Ba-O键连接了14个NMP配体。这是当时聚合度最高的杂金属多钒氧簇合物。
2014年,西班牙的Arriortua课题组合成了一例具有3D结构的{Ni2(H2O)2(Bpa)2}(V6O17)(Bpa=1,2-bis (4-pyridil)ethane)[115]。有趣的是,该化合物在180℃由于Ni2+失去配位的水分子而表现出一个可逆的固态转变过程。高温下的化合物{Ni2(Bpa)2}(V6O17)也保持了结晶的状态,但是这一转化过程包含了1个由单晶到多晶的反应。Ni2+由于失水而空出的配位点由临近的VO4四面体的端O进行补偿,从而稳定了化合物的结构。这类化合物中的结构单元进行重组的机理可以理解为“开关锁”型转化,如图10所示。同一年,南京理工大学的许岩课题组合成了2个基于[V4O12]4-的层状的金属有机单元修饰钒氧簇化合物[Zn(pyim)]2V4O12(pyim=2-(2-pyridyl)imidazole)和[Cu (bim)2]2V4O12(H2O)·CH3CH2OH(bim=bis(1-imidazolyl) methane)[116]并研究了其磁学性能。

图10 通过“关锁”机理生成新Ni-O键示意图[115]Fig.1 0Generation of new Ni-O bonds in the padlock mechanism of{Ni2(H2O)2(Bpa)2}(V6O17)to {Ni2(Bpa)2}(V6O17)
2015年,本课题组利用咪唑类配体合成了三例基于[V4O12]4-钒氧簇化合物[Co2(mIM)5(H2O)2]V4O12(a),[Ni2(mIM)7(H2-O)]V4O12·H2O(b)和[Cd(eIM)2(H2O)] V2O6(c)(mIM=1-methylimidazole,eIM=1-ethylimidazole)。其中化合物[Co2(mIM)5(H2O)2]-V4O12包含1个手性的3D框架结构,在没有手性源的情况下合成手性化合物是比较少见的[155]。同一年,我们通过控制水热反应条件又得到三例Cu甲基咪唑配合物修饰的POVs化合物α-[Cu(mIM)4]V2O6,β-[Cu(mIM)4] V2O6和[Cu(mIM)2)](VO3)2[93]。前2个化合物是一对同分异构体,他们可以通过控制水热反应的温度而分别得到,如图11所示。化合物α-[Cu(mIM)4]V2O6具有一个有趣的3D金刚石互穿拓扑超分子结构。

图11 化合物α-[Cu(mIM)4]V2O6(a),β-[Cu(mIM)4]V2O6(b)和[Cu(mIM)2)](VO3)2(c)的控制合成[93]Fig.1 1Controlling synthesis of compounds α-[Cu(mIM)4]V2O6(a),β-[Cu(mIM)4]V2O6(b)and[Cu(mIM)2)](VO3)2(c)
2 杂多钒氧簇
取代型的POVs是多钒氧簇化合物中一个重要的分支。近年来,主族金属或非金属,过渡金属以及贵金属等都已被引入到多钒氧簇结构中,极大地丰富了多钒氧簇的结构类型。
2.1第一取代的杂多钒氧簇
本课题组于2011年已对取代型的砷-钒氧簇、锑-钒氧簇、硅(锗)-钒氧簇以及硼-钒氧簇合物进行过综述[9],本文仅概括介绍,不再详细赘述。
尽管多钒氧簇合物的结构类型丰富多样,但已报道的非金属(As、Si)或异金属(Sb、Ge)取代的钒氧簇大都是源于十八核钒氧簇{V18O42},它具有由18个VO5四方锥共边连接形成的笼状结构。As第一取代的POVs研究最为广泛,文献众多,在此不再详细列表,有兴趣的读者请参阅相关文献[9,150]。Sb、Si、Ge取代的POVs相对As来说研究的较少,近年来合成的化合物总结请见表4。由这几种元素第一取代的钒氧簇大致可分为以下4个类型:四取代的{α/β-M8V14(X)}[117-119,124-125,131]、三取代的{M6V15(X)}[120-122,126-128]和二取代的{M4V16(X)}[119-124,129](M=As,Sb,Ge,Si),如图12所示。B取代的POVs主要有{V6B20}[133]、{V10B28}[134]、{V12B18}[135-136]、{V12B16}[137]等几个类型簇合物。

图12 多面体/球-棍模型表示的第一取代的阴离子簇之间的转化Fig.1 2Polyhedral/ball-and-stick representation of the polyoxoanion clusters:{V18O42}(a),{α/β-M8V14(X)}(b,c),{M6V15(X)}(d),{M4V16(X)}(e)(M=As, Sb,Ge)
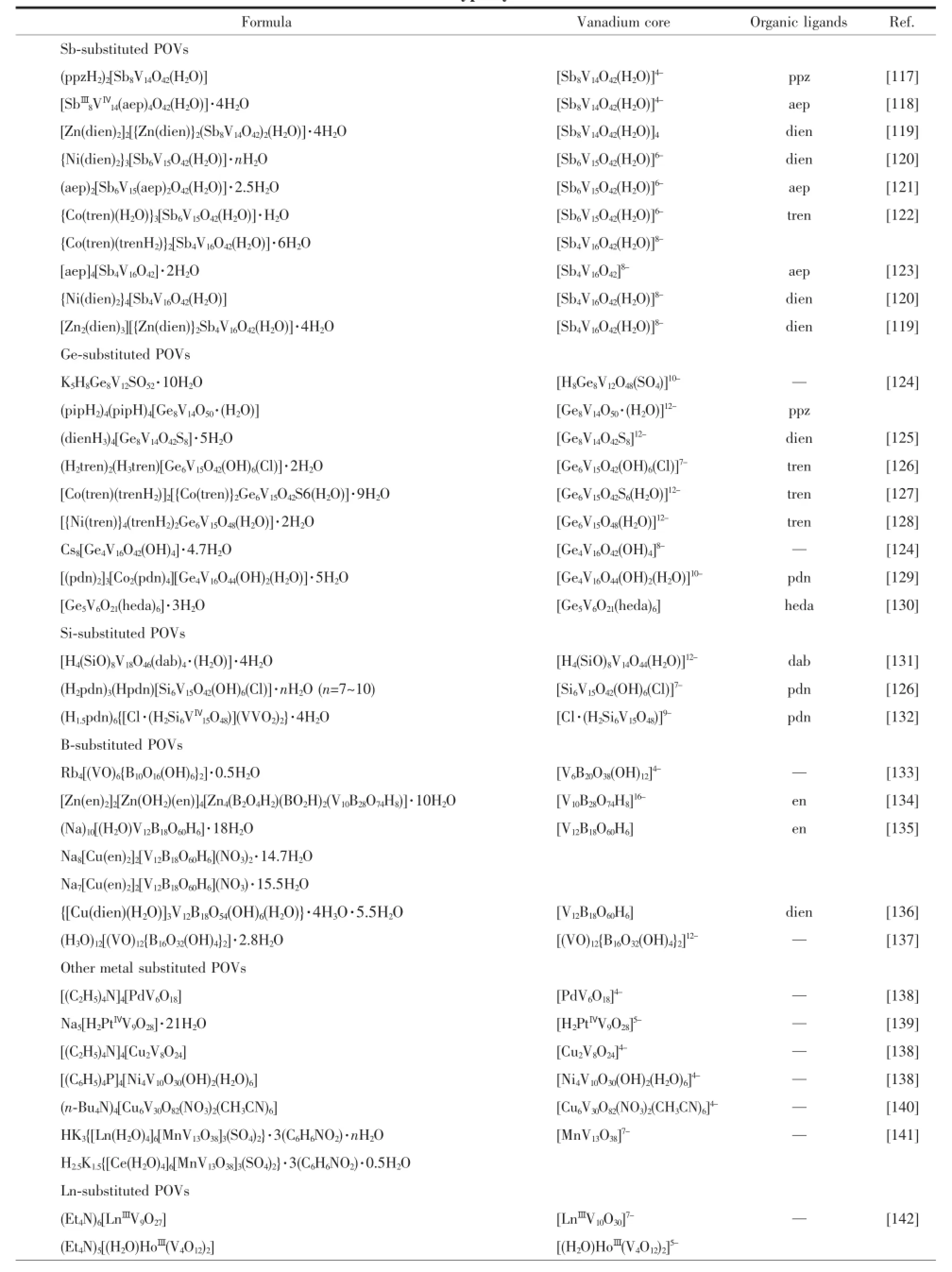
表4 取代型杂多钒氧簇Table 4Substituted-type hybrid POVs clusters

续表4
2012年,苏州大学戴洁课题组合成了一例新型的锗-钒氧簇化合物[Ge5V6O21(heda)6]·3H2O(head=N-(2-hydroxyethyl)ethylenediamine)[130],如图13所示。这是比较少见的不是基于{V18O42}的锗-钒氧簇合物。该簇合物中有VO6、VO5N、GeO4N2八面体,VO5四方锥和GeO4四面体等钒和锗-氧多面体共存。

图13 球-棍(a)和多面体(b~d)模型表示的Ge-POVs阴离子簇Fig.1 3Structure of anion cluster of Ge-POVs in ball and sticks(a)and polyhedron model viewed along different directions(b~d)
由贵金属取代的POVs非常少见,2005年,Hayashi课题组报道了一个新颖贵金属Pd取代的船型结构[(C2H5)4N]4[PdV6O18][138],该结构通过[(C2H5)4N]VO3和[Pd(C6H5CN)2Cl2]在乙腈溶液中反应制得,Pd2+离子通过共价键与V6O18骨架上的端O连接,形成一个船式骨架结构,贵金属Pd2+离子作为船底包覆在整个POVs骨架结构之中。2008年,Lee课题组以四价Pt4+离子和NaVO3为原料合成了一例包覆Pt4+的多钒氧簇化合物Na5[H2PtⅣV9O28]·21H2O[139],在该化合物结构中,Pt4+离子以六配位的方式镶嵌于多钒氧簇骨架中。
近几年,Cu2+、Ni2+、Mn2+等过渡金属以及稀土金属取代的多钒氧簇方面也有不少例子,合成了含有[Cu2V8O24]4-[138],[Cu6V30O82(NO3)2(CH3CN)6]4-[140],[Ni4V10O30(OH)2(H2O)6]4-[138],[MnV13O38]7-[141]以及[LnⅢV9O27]6-,[LnV10O30]7-和[(H2O)HoⅠ(V4O12)2]5-[142]等取代型多钒氧簇阴离子的化合物。
2.2第二取代的杂多钒氧簇
3D过渡金属(TM)引入到POMs体系中的研究主要集中在过渡金属嵌入的钨氧簇合物。而对过渡金属嵌入的钒氧簇合物的研究几乎空白,主要原因是稳定、缺位的钒氧簇前体难以分离。2004年,杨国昱课题组在这一领域取得了突破,他们把Zn2+离子引入到砷钒氧簇中得到了[{Zn(enMe)2}2(enMe)2{Zn2As8V12O40(H2O)}]·4H2O[146]。这也是首次在钒氧簇上实现第二取代反应的例子。随后,他们课题组及王恩波等课题组相继合成了一系列的单金属或双金属取代的TM-As-POVs(TM=Zn2+,Cd2+,Ni2+)[143-147],见图14。
与过渡金属取代的砷钒氧簇相比,过渡金属取代的锗钒氧簇更为稀少,到目前为止,仅有两例报道。2010年,杨国昱课题组合成了第一例基于α-{Ge8V14O50}的2个Cd取代的锗钒氧簇化合物{[(en)2H8Cd2Ge8V12O48][Cd(en)2]2}7H2O[148]。
2014年,同一课题组在此领域的研究又取得最新进展。他们利用水热方法合成了2个4个Cd取代的锗钒氧簇化合物{(CdX)4Ge8VⅣ10O46(H2O)[VⅢ(H2O)2]4(GeO2)4}·8H2O(X=ethylenediamine和1,2-diaminopropane)[149]。这是到目前为止由GeO4四面体和罕见的[VⅢO2(H2O)8]8-连接而成的最大数目的过渡金属取代的锗钒氧簇化合物(图14)。对磁学性质的研究表明,该化合物具有反铁磁性。

图14 多面体/球-棍模型表示的第二取代的阴离子簇之间的转化[143-149]Fig.1 4Polyhedral/ball-and-stick representation of the polyoxoanion clusters
3 多钒氧簇的性质与应用
3.1同多钒氧簇的性质与应用
3.1.1有机官能化POVs的性质与应用
有机官能化的多钒氧簇化合物中一般都存在变价的V,因此,这类化合物也表现出一定的电化学及磁学性质。
德国的Hartl课题组于2003和2005年分别报道了一个甲氧基化的中性的Lindqvist型钒氧簇化合物[VⅤ2VⅣ4O7(OCH3)12][56]。通过电化学测试,证明六个V中有4个+Ⅳ的V,化合物中四价钒核五价钒的比例VⅣ/VⅤ=4∶2。
2012年,本课题组通过调控反应温度和有机配体合成了4个有机砷酸官能化的环形多钒氧簇合物。对化合物[H6VⅤ2VⅣ14O24(OH)8(O3AsC6H4-4-NH2)8]· 4DMF·12H2O的磁性研究表明在VⅣ之间存在强的反铁磁性作用[80]。
有机官能化的多钒氧簇的合成较难,很多都是在无水无氧环境及有机溶剂中合成,这限制了该类材料的广泛研究与应用。有机官能化的多钒氧簇外围的有机组分使得该类化合物更易与有机底物接触,而内部的多钒结构又是很好的催化成分,预期该类化合物在催化有机反应的应用上会有良好的催化效果。近年来,本课题组在这方面做了有益的探索研究工作。
2013年,我们研究了合成的咪唑类配体有机官能化的POVs的催化性能。实验结果表明,此类化合物在以O2为氧化剂条件下,在醇选择性氧化生成醛的反应中表现出较高的催化活性和选择性。同时考察了在相同反应条件下其它钒催化剂,如V2O5、VOSO4和VO(acac)2在苄基烃氧化中的催化性能。其中VⅣ2VⅤ4O14(eIM)8在无需进一步优化的条件下能将水杨醇完全转化,水杨醛的选择性达到96.7%(图15)。重要的是,这些钒氧簇化合物易于回收和重新利用而其催化活性没有改变。在底物扩展实验中,该催化剂对其它几种醇类化合物催化氧化成醛的反应也具有较好的转化率和选择性[87]。

图15 (a)不同催化剂催化在苯甲醇氧化为苯甲醛反应中的催化性能对比;(b)优化条件下VⅣ2VⅤ4O14(eIM)8催化的水杨醇氧化反应[87]Fig.1 5(a)Catalytic properties of different catalysts in the reaction of conversion of benzyl alcohol to benzaldehyde;(b)Oxidation of salicyl alcohol under the preliminary optimized conditions catalyzed by VⅣ2VⅤ4O14(eIM)8
2015年,我们课题组把贵金属Pd配合物为抗衡阳离子的甲氧基化的Lindqvist型钒氧簇化合物应用于苄基烷烃类化合物选择性催化氧化合成苄基酮的反应,如图15所示。发现化合物[Pd(dpa) (acac)]2[VⅤ6O13(OMe)6]作为非均相催化剂,在以叔丁基过氧化氢(TBHP)为氧化剂的条件下,在对氧杂蒽、二苯甲烷、芴、取代芴氧化反应中表现出优异的催化性能,产率最高能达到接近100%。催化剂可完全回收且能重复使用,而催化活性没有明显损失。我们对反应机理的初步探索表明,催化反应中可能存在自由基反应机理,并通过EPR实验得到进一步验证[50]。
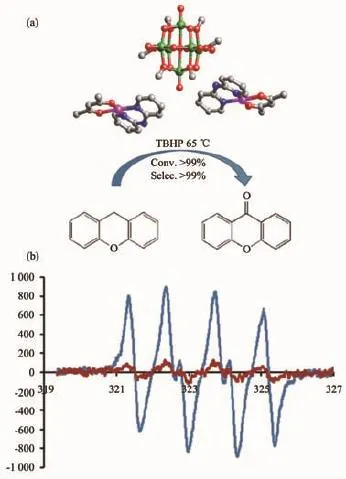
图16 (a)优化条件下[Pd(dpa)(acac)]2[VⅤ6O13(OMe)6]催化的氧杂蒽的选择性氧化反应;(b)二苯甲烷氧化反应中有催化剂(蓝色)和无催化剂(红色)的EPR对比图[50]Fig.1 6(a)Catalytic property of[Pd(dpa)(acac)]2[VⅤ6O13(OMe)6]in the selective oxidation of 9H-xanthene;(b)EPR spectrum with(blue)and without(red)catalyst in the conversion of diphenylmethane
金属有机单元修饰POVs化合物催化性能研究的例子并不多见。2011年,Arriortua课题组将合成杂化的钒酸盐化合物M(C6H16N3)2(VO3)4(M=Co2+,Ni2+,Cu2+)用作硫醚氧化反应的催化剂。实验结果表明,该类化合物在以H2O2或TBHP为氧化剂的条件下能高效、高选择性地将各种硫醚转化为相应的亚砜。图17表示的是以H2O2为氧化剂条件下,催化剂在转化甲基苯基硫醚反应中的动力学曲线。可以看出,所有催化剂在催化行为都比较相似,都能达到83%~64%的转化率和亚砜94%以上的选择性。在催化循环过程中能较好的保持其稳定性和催化活性。这是第一例将此类化合物应用于催化有机反应的例子[151]。

图17 M(C6H16N3)2(VO3)4(M=Co2+,Ni2+,Cu2+)催化的甲基苯基硫醚氧化反应的动力学曲线[151]Fig.1 7Kinetic profile for the oxidation of methyl phenyl sulfide catalyzed by M(C6H16N3)2(VO3)4(M=Co2+, Ni2+,Cu2+)
2013年,同一课题组将[{CoN(H2O)2(Bpe)2}(V4O12)] ·4H2O·Bpe(bpe=1,2-di(4-pyridyl)ethylene)作催化剂,应用于无溶剂条件下醛的硅腈化反应,各种醛的转化率能达到37%~97%[152]。也有将此类催化剂应用于光催化降解有机染料[89,91]及光解水的例子[153]。
2015年,我们研究了甲基咪唑Cu配合物修饰的POVs化合物的催化性能(图18)。实验结果表明,化合物α-[Cu(mIM)4]V2O6能高效、高选择性的催化硫醚的氧化生成相应的亚砜,而作为非均相催化剂,α-[Cu(mIM)4]V2O6能被简便的通过过滤分离,经过多次循环使用而活性没有明显的降低。此化合物在硫醚氧化过程中高效的催化活性也能扩展到不同醇的氧化制备相应的醛的反应,这是第一次将这类化合物应用到醇的选择性催化氧化反应中[93]。
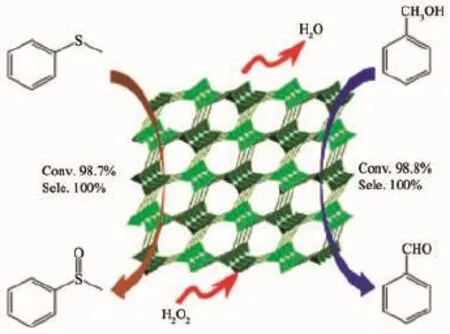
图18 α-[Cu(mIM)4]V2O6催化的甲基苯基硫醚和苯甲醇的选择性氧化反应[93]Fig.1 8Oxidation reaction of methyl phenyl sulfide and benzyl alcohol catalyzed by α-[Cu(mIM)4]V2O6
3.2杂多钒氧簇的性质与应用
杂多钒氧簇由于其它金属或非金属的引入,不仅极大丰富了钒氧簇的结构类型,而且使其具有了在吸附、电化学和磁学等领域更为广泛的应用前景。杂多钒氧簇的相关性质及应用请参考本课题组的中[9]、英文综述[150],在此不再详细讨论。
4 结论与展望
本文综述了近年来在多钒氧簇的修饰及杂化方面取得的研究进展。集中讨论了有机官能化POVs、金属有机单元修饰的POVs以及杂多钒氧簇中的取代型POVs等化合物合成及相关结构,并对各类POVs化合物的性质及应用做了简要介绍。认识到POVs化合物在催化、电化学、光化学以及磁学等领域有着广阔的应用前景,积极开发新型POVs化合物的合成及应用是十分有意义的研究课题。
有关POVs化合物的研究课题,有待从以下两方面进一步展开与深入:
(1)新型POVs化合物的合成。羟基配体官能化的POVs相对苛刻的合成条件及其较差的稳定性增加了该类材料被更广泛研究的难度,设计环境友好、操作简单的合成路线以及引入其它组分以增加其稳定性或许是下一个研究热点。而最近在有机二膦酸及均苯三羧酸官能化多钒氧簇方面的研究表明,采用含有多个活性官能团(如膦酸根,羧基,羟基等)的有机配体修饰POVs,能合成结构多样的有机官能化POVs化合物。在取代型多钒氧簇化合物合成方面,二次取代的多钒氧簇主要集中在过渡金属取代的As-V氧簇,而Ge-V氧簇仅有少数几个例子,Sb-V氧簇和Si-V氧簇则完全空白,这类取代型POVs化合物的合成有很大的扩展空间。
(2)POVs化合物的性能研究。对POVs化合物的性能研究目前主要集中在电化学和磁学领域,而对其在催化(包括有机化合物催化和降解、光解水等)及医药等领域的研究有望成为将来的研究热点之一。期待多钒氧簇化合物的研究不断取得新的进展。
[1]Pope M T.Heteropoly and Isopolyxometalates.Berlin:Springer -verlag,1983:1-10
[2]Hill C L.Chem.Rev.,1998,98:1-2
[3]WANG En-Bo(王恩波),HU Chang-Wen(胡长文),XU Lin (许林).Introduction of Polyoxometalates(多酸化学导论). Beijing:Chemical Industry Press,1997:1-285
[4]Antonova E,Näther C,Kögerler P,et al.Angew.Chem.Int. Ed.,2011,50:764-767
[5]Breen J M,Schmitt W.Angew.Chem.Int.Ed.,2008,47:6904 -6908
[6]Kögerler P,Tsukerblat B,Müller A.Dalton Trans.,2010,39: 21-36
[7]Ichida H,Nagai K,Sasaki Y,et al.J.Am.Chem.Soc.,1989, 111:586-591
[8]Hayashi Y.Coord.Chem.Rev.,2010,255:2270-2280
[9]GAO Yuan-Zhe(高元哲),LI Xiao-Fang(李晓芳),HU Chang-Wen(胡长文).Prog.Chem.(化学进展),2011,23:1050-1059
[10]XU Jia-Ning(徐家宁),YANG Guo-Yu(杨国昱),SUN Hao-Ran(孙浩然),et al.Chin.J.Struct.Chem.(结构化学),1996, 15:458-461
[11]Day V W,Klemperer W G,Yaghi O M.J.Am.Chem.Soc., 1989,111:4518-4519
[12]Klemperer W G.Inorg.Synth.,1992,27:74-85
[13]Evans H T.Inorg.Chem.,1966,5:967-977
[14]XU Jia-Ning(徐家宁),YANG Guo-Yu(杨国昱),SUN Hao-Ran(孙浩然),et al.Chin.J.Struct.Chem.(结构化学),1996, 15:253-256
[15]XU Jia-Ning(徐家宁),YANG Guo-Yu(杨国昱),SUN Hao-Ran(孙浩然),et al.Chin.J.Struct.Chem.(结构化学),1997, 9:576-581
[16]Pvani K,Upreti S,Ramanan A.J.Chem.Sci.,2006,118:159 -164
[17]Humagai H,Arishima M,Kitagawa S.Inorg.Chem.,2002,41: 1989-1992
[18]Liu H X,Wang J,Jian F F,et al.J.Clust.Sci.,2009,20:621-627
[19]Putrevu N R,Doedens R J,Khan M I.Inorg.Chem.Comm., 2013,38:5-7
[20]Sánchez-Lombardo I,Sánchez-Lara E,Pérez-Benítez A, et al.Eur.J.Inorg.Chem.,2014:4581-4588
[21]Lyer A K,Roy S,Haridasan R,et al.Dalton Trans.,2014, 43:2153-2160
[22]Day V W,Klemperer W G,Yaghi O M.J.Am.Chem.Soc., 1989,111:5959-5961
[23]Hou D,Hagen K S,Hill C L.J.Am.Chem.Soc.,1992,114: 5864-5866
[24]Chen L,Jiang F,Lin Z,et al.J.Am.Chem.Soc.,2005,127: 8588-8589
[25]Müller A,Penk M,Rohljing R,et al.Angew.Chem.Int. Ed.,1990,29:926-927
[26]Karet G B,Sun Z M,Streib W E,et al.Chem.Commun., 1999:2249-2250
[27]Long D L,Orr D,Seeber G,et al.J.Clust.Sci.,2003,14: 312-324
[28]Khan M I,Ayesh S,Doedens R J,et al.Chem.Commun., 2005:4658-4660
[29]Müller A,Krickemeyer E,Penk M,et al.Angew.Chem.Int. Ed.,1991,30:1674-1677
[30]Suber L,Bonamico M,Fares V.Inorg.Chem.,1997,36:2030 -2033
[31]Müller A,Penk M,Krickemeyer E,et al.Angew.Chem.Int. Ed.,1988,27:1719-1721
[32]Müller A,Rohlfing R,Doring J,et al.Angew.Chem.Int. Ed.,1991,30:588-590
[33]Wang Y,Yu J H,Pan Q H,et al.Inorg.Chem.,2004,43: 559-565
[34]Qi Y J,Wang Y H,Li H M,et al.J.Mol.Struct.,2003,650: 123-129
[35]Zhang X,You W S,Zhu Z M,et al.Inorg.Chem.Comm., 2006,9:526-528
[36]Zheng L M,Wang Y S,Wang X Q,et al.Inorg.Chem., 2001,40:1380-1385
[37]Li Y G,Lu Y,Luan G Y,et al.Polyhedron,2002,21:2601-2608
[38]Dobrick M S,Jansen M.Inorg.Chem.,2007,46:4380-4382
[39]Done B X,Gómez-García C J,Peng J,et al.Polyhedron, 2007,26:1310-1316
[40]Yi Z H,Yu X Y,Xia W J,et al.CrystEngComm,2010,12: 242-249
[41]Lu Y,Wang E B,Yuan M,et al.J.Mol.Struct.,2002,607: 189-194
[42]La Duca R L,Brodkin C,Finn R C,et al.Inorg.Chem. Commun.,2000,3:248-250
[43]Law T S C,Sung H H Y,Williams I D,et al.Inorg.Chem. Commun.,2000,3:420-423
[44]La Duca R L,Rarig R S,Zubieta J.Inorg.Chem.,2001,40: 607-612
[45]Law T S C,Williams I D.Chem.Mater.,2000,12:2070-2072
[46]Hagrman P J,Bridges C,Greedan J E,et al.Dalton Trans., 1999,28:2901-2903
[47]Khan M I,Zubieta J.Progress in Inorganic Chemistry.New York:John Wiley&Sons Inc,1995.
[48]Dolbecq A,Dumas E,Mayer C R,et al.Chem.Rev.,2010, 110:6009-6048
[49]Piepenbrink M,Triller M U,Gorman N H J,et al.Angew. Chem.,Int.Ed.,2002,41:2523-2525
[50]Li J K,Huang X Q,Yang S,et al.Inorg.Chem.,2015,54: 1454-1461
[51]Hou D,Kim G S,Hagen K S,et al.Inorg.Chim.Acta,1993, 211:127-130
[52]Adach A,Daszkiewicz M,Cieslak-Golonka M.Polyhedron, 2012,47:104-111
[53]Daniel C,Hartl H.J.Am.Chem.Soc.,2009,131:5101-5114
[54]Kessler V G,Seisenbaeva G A.Inorg.Chem.Commun., 2000,3:203-204
[55]Spandl J,Daniel C,Brüdgam I,et al.Angew.Chem.Int. Ed.,2003,42:1163-1166
[56]DanielC,HartlH.J.Am.Chem.Soc.,2005,127:13978-13987
[57]Chen Q,Liu S C,Zubieta J.Eur.J.Inorg.Chem.,1989,28: 4433-4434
[58]Chen Q,Zubieta J.Inorg.Chem.,1990,29:1456-1458
[59]Chen Q,Goshorn D P,Scholes C P,et al.J.Am.Chem. Soc.,1992,114:4667-4681
[60]Khan M I,Chen Q,Goshorn D P,et al.Inorg.Chem.,1992, 31:1556-1558
[61]Khan M I,Chen Q,Hpe H,et al.Inorg.Chem.,1993,32: 2929-2937
[62]Batchelor L J,Shaw R,Markey S J,et al.Chem.Eur.J., 2010,16:5554-5557
[63]Müller A,Meyer J,Bgge H,et al.Z.Anorg.Allg.Chem., 1995,621:1818-1831
[64]Yin P C,Wu P F,Xiao Z C,et al.Angew.Chem.Int.Ed., 2011,50:2521-2525
[65]Han J W,Hill C L.J.Am.Chem.Soc.,2007,129:15094-15095
[66]Santoni M P,Pal A K,Hanan G S,et al.Inorg.Chem.,2011, 50:6737-6745
[67]Wu P F,Xiao Z C,Zhang J,et al.Chem.Commun.,2011, 47:5557-5559
[68]Khan M I,Chen Q,Zubieta J.Chem.Commun.,1992:305-306
[69]Khan M I,Chen Q,Goshorn D P,et al.J.Am.Chem.Soc., 1992,114:3341-3346
[70]Khan M I,Lee Y S,O′Connor C J,et al.J.Am.Chem.Soc., 1994,116:5001-5002
[71]Aronica C,Chastanet G,Zueva E,et al.J.Am.Chem.Soc., 2008,130:2365-2371
[72]Tidmarsh I S,Laye R H,Brearley P R,et al.Chem.Eur.J., 2007,13:6329-6338
[73]Tidmarsh I S,Laye R H,Brearley P R,et al.Chem.Commun., 2006:2560-2562
[74]Spandl J,Brüdgam I,Hart H,et al.Angew.Chem.,Int.Ed., 2001,40:4018-4020
[75]Barthelet K,Riou D,Férey G.Chem.Commun.,2002:1492 -1493
[76]Zhang Z J,Wojtas L,Zaworotko J.Chem.Sci.,2014,5:927-931
[77]Breen J M,Schmitt W.Angew.Chem.,Int.Ed.,2008,47: 6904-6908
[78]Breen J M,Clérac R,Zhang L,et al.Dalton Trans.,2012, 41:2918-2926
[79]Zhang L,Schmitt W.J.Am.Chem.Soc.,2011,133:11240-11248
[80]Chen B K,Wang B,Lin Z G,et al.Dalton Trans.,2012,41: 6910-6913
[81]Rabeah J,Ster R,Jiao H,et al.Chem.Eur.J.,2012,18: 6433-6436
[82]Konar S,Clearfield A.Inorg.Chem.,2008,47:3492-3494
[83]Ushak S,Spodine E,Fur E L,et al.Inorg.Chem.,2006,45: 5393-5398
[84]Mahimaidoss M B,Krasnikov S A,Reck L,et al.Chem. Commun.,2014,50:2265-2267
[85]Duan C Y,Tian Y P,Lu Z L,et al.Inorg.Chem.,1995,34: 1-2
[86]Fu M L,Guo J C,Wu A Q,et al.Eur.J.Inorg.Chem.,2005: 3104-3108
[87]Chen B K,Huang X Q,Wang B,et al.Chem.Eur.J.,2013, 19:4408-4413
[88]Wang X,Chen B,Liu G,Lin H,et al.J.Inorg.Organomet. Polym.Mater.,2009,19:176-180
[89]Hu Y,Luo F,Dong F.Chem.Commun.,2011,47:761-763
[90]Zhang C D,Liu S X,Xie L H,et al.J.Mol.Struct.,2005, 753:40-44
[91]Lin H,Maggard P A.Inorg.Chem.,2008,47:8044-8052
[92]Luis R F,Urtiaga M K,Mesa J L,et al.CrystEngComm, 2011,13:6488-6498
[93]Li J K,Huang X Q,Yang S,et al.Cryst.Growth Des.,2015, 15:1907-1914
[94]DeBurgomaster P,Zubieta J.Inorg.Chim.Acta,2010,363: 2912-2919
[95]Qi Y F,Lü C P,Li Y G,et al.Inorg.Chem.Commun.,2010, 13:384-387
[96]Zhang X M,Tong M L,Chen X M.Chem.Commun.,2000, 36:1817-1818
[97]Thomas J,Agarwal M,Ramanan A,et al.CrystEngComm, 2009,11:625-631
[98]Gao G G,Cheng P S,Mak T C W.J.Am.Chem.Soc.,2009, 131:18257-18259
[99]Lan Y Q,Li S L,Su Z M,et al.Chem.Commun.,2008,44: 58-60
[100]Kastner K,Puscher B,Streb B.Chem.Commun.,2013,49: 140-142
[101]Zhou J,Liu X,Hu F L,et al.CrystEngComm,2013,15: 4593-4596
[102]Teng Y L,Dong B X,Peng J,et al.CrystEngComm,2013, 15:2783-2785
[103]Zhang Z B,Xu Y,Zheng L,et al.CrystEngComm,2011, 13:2191-2193
[104]Wang X L,Chen B K,Liu G C,et al.J.Organomet.Chem., 2010,695:827-832
[105]Ellsworth J M.,Smith M D,zur Loye H C.Solid State Sci., 2008,10:1822-1834
[106]Zheng L M,Wang X,Wang Y,et al.J.Mater.Chem.,2001, 11:1100-1105
[107]Zheng L M,Whitfield T,Wang X,et al.Angew.Chem., Int.Ed.,2000,39:4528-4531
[108]Rarig Jr R S,Zubieta J,et al.Dalton Trans.,2003:1861-1868
[109]Lin B Z,Liu X S.J.Chem.Soc.,Dalton Trans.,2002,31: 865-869
[110]Maggard P A,Boyle P D.Inorg.Chem.,2003,42:4250-4252
[111]Cui X B,Lin Z E,Yang G Y.Solid State Sci.,2003,5:311-315
[112]Yan B B,Maggard P A.Inorg.Chem.,2007,46:6640-6646
[113]Maggard M I K,Yohannes E,Golub V O,et al.Chem. Mater.,2007,19:4890-4895
[114]Kastner K,Puscher B,Streb C.Chem.Commun.,2013,49: 140-142
[115]de Luis R F,Orive J,Larrea E S,et al.Cryst.Growth Des., 2014,14:658-670
[116]Hou W,Guo J,Xu X,et al.Dalton Trans.,2014,43:865-871
[117]Antonova E,Wutkowski A,Nther C,et al.Solid State.Sci., 2011,13:2154-2159
[118]Antonava E,Nther C,Kgerler P,et al.Angew.Chem.,Int. Ed.,2011,50:764-767
[119]Gao Y Z,Han Z G,Xu Y Q,et al.J.Cluster Sci.,2010,21:163-171
[120]Antonava E,Nther C,Bensch W.Dalton Trans.,2012,41: 1338-1344
[121]Antonava E,Seidlhofer B,Wang J,et al.Chem.Eur.J., 2012,18:15316-15322
[122]Antonava E,Nther C,Bensch W.CrystEngComm,2012,14: 6853-6859
[123]Kiebach R,Nther C,Bensch W.Solid State Sci.,2006,8: 964-970
[124]Whitfield X,Wang X,Jacobson A J.et al.Inorg.Chem., 2003,42:3728-3733
[125]Pitzschke D,Wang J,Hoffmann R D,et al.Angew.Chem., Int.Ed.,2006,45:1305-1308
[126]Gao Y Z,Xu Y Q,Li S,et al.J.Coord.Chem.,2010,63: 3373-3383
[127]Wang J,Nther C,Kgerler P,et al.Eur.J.Inorg.Chem., 2012:1237-1242
[128]Wang J,Nther C,Spldrich M,et al.CrystEngComm,2013, 15:10238-10245
[129]Gao Y Z,Xu Y Q,Huang K L,et al.Dalton Trans.,2012, 41:6122-6129
[130]You L S,Zhu Q Y,Zhang X,et al.CrystEngComm,2013, 15:2411-2415
[131]Tripathi A,Hughbanks T,Clearfield A.J.Am.Chem.Soc., 2003,125:10528-10529
[132]Gao Y Z,Xu Y Q,Cao Y,et al.Dalton Trans.,2012,41: 567-571
[133]Warren C J,Rijssenbeek J T,Rose D J,et al.Polyhedron, 1998,17:2599-2605
[134]Wu M M,Law T S C,Sung H H Y,et al.Chem.Commun., 2005:1827-1829
[135]Brown K,Car P E,Vega A,et al.Inorg.Chim.Acta,2011, 367:21-28
[136]Zhou J,Liu X,Chen R,et al.CrystEngComm,2013,15: 5057-5063
[137]Warren C J,Rose D J,Haushalter R C,et al.Inorg.Chem., 1998,37:1140-1141
[138]Kurata T,Uehara A,Hayashi Y,et al.Inorg.Chem.,2005, 44:2524-2530
[139]Lee U,Joo H C,Park K M,et al.Angew.Chem.Int.Ed., 2008,47:793-796
[140]Forster J,Rsner B,Fink R H,et al.Chem.Sci.,2013,4: 418-424
[141]Liu D,Lu Y,Li Y G,et al.Dalton Trans.,2013,42:14445-14453
[142]Nishio M,Inami S,Katayama M,et al.Inorg.Chem.,2012, 51:784-793
[143]Cui X B,Xu J Q,Meng H.Inorg.Chem.,2004,43:8005-8009
[144]Zheng S T,Zhang J,Yang G Y.Inorg.Chem.,2005,44: 2426-2430
[145]Zheng S T,Wang M H,Yang G Y.Inorg.Chem.,2007,46: 9503-9508
[146]Zheng S T,Zhang J,Yang G Y.Eur.J.Inorg.Chem.,2007, 46:2004-2007
[147]Qi Y F,Li Y G,Qin C.Inorg.Chem.,2007,46:3217-3230
[148]Zhou J,Zhang J,Fang W H,et al.Chem.Eur.J.,2010,16: 13253-13261
[149]Zhou J,Zhao J W,Wei Q,et al.J.Am.Chem.Soc.,2014, 136:5065-5071
[150]Gao Y Z,Chi Y N,Hu C W.Polyhedron,2014,83:242-258
[151]Larrea E,Mesa J L,Pizarro J L,et al.Dalton Trans.,2011, 40:12690-12698
[152]de Luis R F,Urtiaga M K,Mesa J L,et al.Inorg.Chem., 2013,52:2615-2626
[153]LuoL,MaggardPA.Cryst.GrowthDes.,2013,13:5282-5288
[154]de Luis R F,Orive J,Larrea E S,et al.CrystEngComm, 2014,16:10332-10366
[155]Wu S J,Yang X H,Hu J F,et al.CrystEngComm,2015, 17:1625-1630
Progress in Polyoxovanadate Chemistry
LI Ji-Kun1,2HU Chang-Wen*,1
(1Key Laboratory of Cluster Science,Ministry of Education,School of Chemistry and Beijing Institute of Technology,Beijing 100081,China)
(2College of Chemistry and Chemical Engineering,Taishan University,Tai′an,Shandong 271021,China)
Due to the advantages of their regulated composition,structure and size,redox activity and low corrosivity,etc.,polyoxovanadium clusters have broad application prospects in the field of optical,magnetic, catalytic,medicine and become one of the hot research topics of polyoxometalates chemistry.Based on the recent investigations,this paper summarizes the related advancements in the synthesis,structure and properties of polyoxovanadium clusters and gives their researching prospects.
polyoxovanadium clusters;organofunctionalized;hybrid;properties;application
O614.51+1
A
1001-4861(2015)09-1705-21
10.11862/CJIC.2015.247
2015-05-30。收修改稿日期:2015-07-13。
国家自然科学基金(No.21173021,21231002和21276026),111课题(No.B07012),973项目(No.2014CB932103)和山东省自然科学基金(No.ZR2013BL012)资助项目。
*通讯联系人。E-mail:cwhu@bit.edu.cn;会员登记号:S06N8447M1305。
猜你喜欢
英语文摘(2022年8期)2022-09-02
当代化工研究(2022年12期)2022-07-11
英语文摘(2021年1期)2021-06-11
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
当代陕西(2019年6期)2019-04-17
中文信息(2018年1期)2018-03-22
固体火箭技术(2017年5期)2017-11-06
传媒评论(2017年12期)2017-03-01
信息记录材料(2016年4期)2016-03-11
中国塑料(2015年8期)2015-10-14
