
p38 MAPK/p53信号通路调控骨肉瘤细胞中Etheràgo-go表达的研究
2015-11-04 07:18吴进刘庆军陈志达曾文容吴欣宇林斌
中国癌症防治杂志 2015年5期
吴进刘庆军陈志达曾文容吴欣宇林斌
作者单位:363000 漳州 厦门大学附属东南医院1骨科,2神经内科
基础研究
p38 MAPK/p53信号通路调控骨肉瘤细胞中Etheràgo-go表达的研究
吴进1刘庆军1陈志达1曾文容1吴欣宇2林斌1
作者单位:363000漳州厦门大学附属东南医院1骨科,2神经内科
目的 检测Etheràgo-go(Eag)在人骨肉瘤细胞中的表达并探索其调控的分子机制。方法 采用实时定量逆转录-聚合酶链反应(reverse transcription polymerase chain reaction,RT-PCR)和免疫印迹技术(Western blot,WB)检测骨肉瘤细胞MG-63中Eag的表达。体外实验检测Eag抑制剂对MG-63细胞增殖的影响,体内实验检测Eag短发卡RNA(short hairpin RNA,shRNA)对裸鼠骨肉瘤生长的影响。最后用WB检测骨肉瘤细胞中促有丝分裂原活化蛋白激酶(mitogenactivated protein kinase,MAPK)和p53蛋白的表达水平。 结果 Eag在MG-63细胞中高表达,Eag shRNA或抑制剂丙咪嗪(Imipramine)能从体内外有效地抑制人骨肉瘤细胞增殖。p38 MAPK抑制剂SB203580或小干扰RNA(small interference RNA,siRNA)可抑制MG-63细胞的增殖,同时诱导p53的表达。p53激活剂nutlin-3可抑制MG-63细胞增殖并下调Eag的表达,而p53抑制剂pifithrin-alpha(PFT-α)则能促进MG-63细胞增殖并诱导Eag的表达。结论Eag作为癌基因参与了骨肉瘤细胞增殖过程,其可能受p38MAPK/p53信号通路的调控。
骨肿瘤;Etheràgo-go通道;细胞增殖;MAPK信号通路;p53基因;调控;表达
骨肉瘤是青少年最常见的恶性骨肿瘤,其恶性程度高,容易复发和转移,严重影响青少年的生命健康[1]。即使采用外科手术并联合新辅助化疗的标准方案治疗,无肺部转移患者的5年生存率仅为65%,如发生肺转移患者的预后更差[2,3]。因此深入研究与骨肉瘤发生、发展和治疗相关的分子机制,成为目前研究骨肉瘤较为关注的疑难问题,也是如何延长骨肉瘤患者生存时间的关键。
Etheràgo-go(Eag)及其编码的Eag通道是第一个被证明与肿瘤密切相关的钾离子通道[4]。近年来,Eag已成为肿瘤领域研究的热点并不断有新进展[4,5]。但其在骨肉瘤中的作用及机制尚未明确。本研究首先通过PT-PCR和WB法检测Eag在人骨肉瘤细胞中的表达,之后通过腺病毒感染抑制Eag的表达并在体外水平验证其在调控MG-63细胞增殖中的作用,并初步阐明其调控机制。
1 材料与方法
1.1实验材料
1.1.1细胞人骨肉瘤细胞株MG-63、人成骨细胞瘤细胞株hFOB 1.19、人黑色素瘤细胞株MDA-MB435S和人胚肾细胞株HEK293均购自美国ATCC公司。
1.1.2主要试剂DMEM培养基、RPMI-1640培养基、胎牛血清购自美国Gibco公司;细胞培养板、丙咪嗪、二甲基亚砜(DMSO)、JNK MAPK通道阻滞剂CEP11004购自美国Sigma公司;CCK-8试剂盒购自美国Dojindo公司;p38 MAPK通道阻滞剂SB203580购自德国Darmstadt公司;ERK1/2 MAPK通道抑制剂PD98059、p53通道激活剂nutlin-3、p53通道抑制剂PFT-α、辣根过氧化物酶偶联山羊抗兔IgG抗体和Eag siRNA购自美国SantaCruz公司;p38MAPK siRNA、JNK MAPK siRNA、ERK1/2MAPK siRNA和controlsiRNA购自美国Cell Signaling Technology公司;Eag单克隆抗体购自以色列Alomone公司;增强化学发光(ECL)显色试剂盒购自美国Millipore公司;细胞裂解液购自美国Invitrogen公司;反转录试剂盒购自日本Takara公司。
1.2实验方法
1.2.1细胞培养及转染MG-63、MDA-MB435S和HEK293细胞培养于RPMI-1640培养基(含10%胎牛血清、100 U/ml青霉素和100 mg/L链霉素),hFOB 1.19细胞培养于F12/DMEM培养基(含10%胎牛血清、100 U/ml青霉素和100mg/L链霉素),均置于37℃、5%CO2孵育箱中培养及传代。转染前1 d,取处于对数生长期的MG-63细胞(1×105个/ml)接种于6孔板中,待细胞密度达80%时,使用笔者等[6]之前构建的腺病毒表达载体Ad5-Eag-shRNA和Ad5-Control-shRNA进行细胞感染,8 h后换液,继续孵育48 h。
1.2.2蛋白印迹技术收集(5~6)×107个对数生长期细胞,用4℃预冷的细胞裂解缓冲液(10mmol/LEDTA、10mmol/L Tris-HCl、150mmol/LNaCl、0.4%SDS、pH 7.4)裂解细胞,摇晃充分混匀细胞。然后移入离心管,在4℃离心半径8 cm下15 000 r/min离心约10min,吸取上清液,用二锌可酸(bicinchonininc acid,BCA)法测定蛋白浓度后置于-80℃冰箱保存。上样前把样品置于沸水浴中5~8 min,然后在100V电流下进行12% SDS-聚丙烯酰胺凝胶电泳(SDS-polyacrylamide gel electrophoresis,SDS-PAGE)2 h分离蛋白,用电转膜仪30伏湿法转膜过夜(转膜前用硝酸纤维素膜转移缓冲液浸泡30min)。用含10%脱脂奶粉的磷酸盐吐温缓冲液盐水(phosphate buffered saline tween,PBST)室温封闭1.5 h,弃去封闭液,然后在室温下分别用Eag、β-actin、p-ERK1/2、ERK1/2、p-JNK、JNK、p-p38 MAPK、p38 MAPK和p53抗体孵育,置于4℃冰箱过夜;用PBS洗涤3次,每次10 min,加入相对应的二抗室温孵育1.5 h,1∶1 000稀释;用PBS洗涤3次,每次10 min;采用ECL显色法检测,内参为β-actin,用凝胶成像分析软件分析条带净灰度,用各组目的蛋白的灰度值除以β-actin的灰度值以校正误差,所得结果为目的蛋白的相对含量,表示目的蛋白的表达水平。
1.2.3RT-PCR用 Trizol法提取各组细胞的总RNA,按照反转录试剂盒说明书的步骤制备cDNA,引物序列如下:Eag上游引物为5′-GCTTTTGAGAACGTGGATGAG-3′,下游引物为5′-CGAAGATGGTGGCATAGAGAA-3′,475 bp,56℃;β-actin上游引物为5′-TCCACCTTCCAGCAGATGTG-3′,下游引物为5′-GCATTTGCGGTGGACGAT-3′,75 bp,54℃。反应体系:0.5μlcDNA,7.5μl超纯水,上下游引物各1.0μl,SYBR Green 10.0μl,共计20μl。反应条件:⑴预变性:94℃,4min;⑵变性:94℃,45 s;⑶退火:54℃,45 s;⑷延伸:72℃,45s,循环30次。
1.2.4CCK-8法将1×105个细胞置于96孔板中,每孔设置3个副孔。无血清RPMI-1640培养液培养12 h后分别处理,继续培养48 h后收集细胞。10ml CCK-8溶液加入每孔中孵育1 h,选择450 nm波长,在酶联免疫监测仪上测定各孔光吸收值。
1.2.5建立裸鼠骨肉瘤异种移植模型选用6~8周无胸腺的BALB/c雌性裸鼠作为实验对象,严格按照实验动物伦理规定进行实验操作。根据笔者之前的方法[6],通过皮下注射l50μl(1.5×106个/ml)MG-63细胞悬液在裸鼠右侧后腿诱导肿瘤,1周后可长出肉眼可见的肿瘤。之后将裸鼠分为3组(每组6只),实验组:接受瘤内注射Ad5-Eag-shRNA(10 MOI),2 d/次,共6次;阴性对照组:接受瘤内注射Ad5-ControlshRNA(10 MOI),2 d/次,共6次;空白对照组:接受瘤内生理盐水注射(10m l),2 d/次,共6次。每2 d测量肿瘤体积,收集结果,绘制生长曲线图。体积计算的参考公式:1/2×L×W2,L为肿瘤长度,W为肿瘤宽度。
1.3统计学方法
采用SPSS 18.0软件对结果进行统计分析。数据以均数±标准差(x±s)表示,采用t检验或单因素方差分析(ANOVA)。以P<0.05为差异有统计学意义。
2 结果
2.1Eag在骨肉瘤细胞中的表达
RT-PCR检测结果示:Eag在MG-63细胞中与阳性对照组MDA-MB435S细胞中均为高表达,且明显高于阴性对照组hFOB 1.19细胞(图1A)。WB检测进一步证实了Eag在骨肉瘤细胞中异常高表达(图1B)。
2.2Eag沉默对骨肉瘤细胞增殖的影响
图2A显示:Ad5-Eag-shRNA可有效抑制MG-63细胞中Eag的表达。与空白对照组相比,Ad5-Eag-shRNA[(48.55±3.07)%,图2B]和丙咪嗪[(56.17±3.22)%,图2C]均可明显抑制MG-63细胞增殖,差异有统计学意义(P均<0.001)。
2.3Eag沉默对裸鼠骨肉瘤生长的影响
图3显示:从第10天起,实验组裸鼠骨肉瘤体积小于空白对照组和阴性对照组,差异有统计学意义(P均<0.01)。与空白对照组和阴性对照组相比,实验组可有效地抑制裸鼠骨肉瘤的生长。
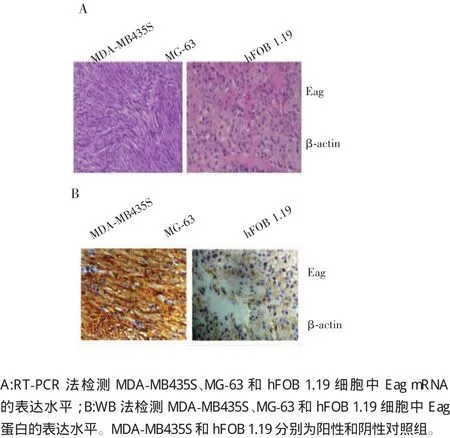
图1 Eag在骨肉瘤细胞中的表达
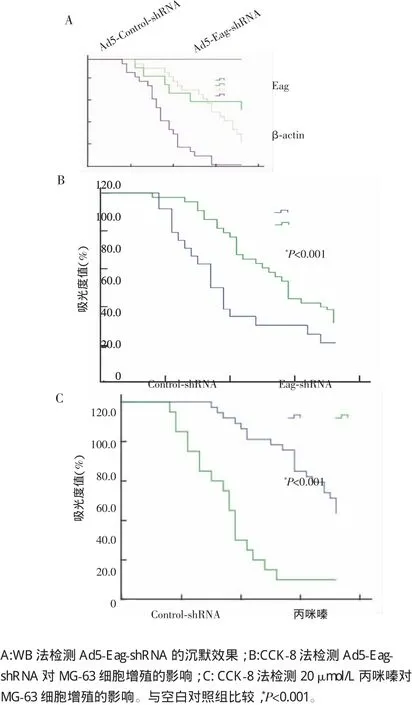
图2 Eag沉默对MG-63细胞增殖的影响

图3 Eag沉默对裸鼠骨肉瘤生长的影响
2.4p38MAPK通路的活化促进骨肉瘤细胞增殖
WB法检测结果示:MG-63细胞中总p38 MAPK和磷酸化p38MAPK的表达水平明显高于其在hFOB 1.19细胞中的表达水平。而总JNK MAPK、ERK1/2 MAPK和磷酸化JNK MAPK、ERK1/2 MAPK在两株细胞中表达无明显异常(图4A)。CCK-8法实验显示:与空白对照组相比,p38 MAPK抑制剂SB203580和p38 MAPK siRNA均可明显抑制MG-63细胞增殖,差异均具有统计学意义(P均<0.001)。而JNK MAPK抑制剂CEP11004、JNK MAPK siRNA、ERK1/2 MAPK抑制剂PD98059和ERK1/2 MAPK siRNA对MG-63细胞增殖均无明显影响(图4B和图4C)。

图4 MG-63细胞中p38MAPK信号通路激活并促细胞增殖
2.5p38 MAPK/p53信号通路调控MG-63细胞中Eag通道的表达
WB法检测结果示:p38 MAPK抑制剂SB203580和p38 MAPK siRNA可分别诱导MG-63细胞中p53的表达并抑制Eag的表达(图5A)。p53激活剂nutlin-3可抑制MG-63细胞增殖(图5B)并下调Eag的表达(5C),而p53抑制剂PFT-α则能促进MG-63细胞增殖(图5D)并诱导Eag的表达(图5E),促细胞增殖的效应可被Ad5-Eag-shRNA部分抵消(图5D)。

图5 MG-63细胞中p38MAPK/p53信号通路调控Eag的表达
3 讨论
研究表明,Eag基因在人类正常组织中只表达于脑组织、胎盘,短暂性表达于肌原细胞,异常表达于多种人类肿瘤细胞系和肿瘤组织[7],Eag在正常组织和相对应的肿瘤组织中表达的差异性使其成为研究的热点。本研究的RT-PCR和WB检测结果证实Eag在骨肉瘤与成骨细胞中存在差异性表达,为后续进一步研究Eag与骨肉瘤的关系奠定了基础。丙咪嗪是一种常见的抗抑郁药物,但其特异性差,对心肌细胞上的某些离子通道有较强的抑制作用[8]。有研究[8]报道,包括丙咪嗪在内的多种Eag抑制剂均可达到抑制肿瘤细胞增殖的效果。相对于丙咪嗪,siRNA具有更高的特异性,而且无明显副作用[9]。Eag基因编码的产物为Eag钾离子通道,是一个与肿瘤密切相关的钾离子通道,在其促进肿瘤发生的过程中不仅仅表现出了离子通道的特性,当利用特殊的药物废除Eag的通道电流,Eag仍能表现出促肿瘤发生的能力,这也是其最吸引研究人员的特性[10]。本实验结果显示,Eag非特异性抑制剂丙咪嗪抑制骨肉瘤增殖是通过阻断Eag离子通道传导,但Eag-siRNA在体内外水平有效地抑制骨肉瘤增殖的机制则不依赖于Eag的离子通道特性,提示Eag基因与骨肉瘤的发生、发展密切相关。
前期的研究证实Eag基因参与了多种肿瘤发生、发展的过程,但其具体的调控机制尚不明确。MAPK信号通路是一类丝氨酸/苏氨酸蛋白激酶,能够将细胞外信号传导至细胞核内,调节相关基因表达,从而调控细胞增殖、周期、分化等诸多过程。近年来研究显示MAPK通路参与了包括骨肉瘤在内的多种肿瘤的发生、发展过程[11,12]。结合本研究的结果,即在骨肉瘤中异常高表达的Eag可激活p38 MAPK通路,从而实现促骨肉瘤细胞增殖的表型,而抑制p38 MAPK则可阻滞骨肉瘤细胞增殖,提示p38 MAPK的活化与Eag异常高表达密切相关,由此推测,位于细胞核周围的Eag可激活MAPK信号通道从而促进肿瘤细胞增殖[8],但其具体的调控机制仍需进一步阐明。
Lin等[13]发现在人类成神经细胞瘤SH-SY5Y细胞中,p53通过p53-miR34-E2F1信号通路负反馈调控Eag。本研究也得到了类似的结果,即p38 MAPK抑制剂SB203580或siRNA可诱导骨肉瘤细胞中p53的表达,提示p53可能是p38 MAPK的下游因子。此外,通过p53的激活剂或抑制剂改变p53的表达水平可诱导Eag表达水平的变化。综上所述,在骨肉瘤中,p38 MAPK和Eag形成了一个正反馈循环,即Eag高表达可激活p38 MAPK,抑制p53的表达,而上调p53的表达则可抑制Eag表达。
本研究证实了Eag作为癌基因参与了骨肉瘤细胞增殖过程,其可能受p38 MAPK/p53信号通路调控,抑制p38 MAPK或Eag的表达有望成为一种新的骨肉瘤治疗方案。
[1] 王显阳,常君丽,施杞,等.骨肉瘤相关信号通路的研究进展[J].中国癌症防治杂志,2015,7(1):52-55.
[2] Endo-Munoz L,Evdokiou A,Saunders NA.The role of osteoclasts and tumour-associated macrophages in osteosarcoma metastasis[J]. Biochim Biophys Acta,2012,1826(2):434-442.
[3] Luetke A,Meyers PA,Lewis I,et al.Osteosarcoma treatment-where do westand?A stateof theart review[J].Cancer TreatRev,2014,40(4):523-532.
[4]Pardo LA,del Camino D,Sánchez A,et al.Oncogenic potential of EAG K(+)channels[J].EMBO J,1999,18(20):5540-5547.
[5] Haitin Y,Carlson AE,Zagotta WN.The structural mechanism of KCNH-channel regulation by the eag domain[J].Nature,2013,501(7467):444-448.
[6] Wu J,Wu X,ZhongD,etal.Shorthairpin RNA(shRNA)Etheràgo-go1(Eag1)inhibition of human osteosarcoma angiogenesis via VEGF/ PI3K/AKT signaling[J].Int JMol Sci,2012,13(10):12573-12583.
[7] Camacho J.Etheràgo-go potassium channels and cancer[J].Cancer Lett,2006,233(1):1-9.
[8] Asher V,Sowter H,Shaw R,et al.Eag and HERG potassium channels as novel therapeutic targets in cancer[J].World JSurg Oncol,2010,8:113.
[9]Weber C,Mello de Queiroz F,Downie BR,et al.Silencing the activity and proliferative properties of the human EagIPotassium Channel by RNA Interference[J].JBiol Chem,2006,281(19):13030-13037.
[10]Downie BR,Sánchez A,Knötgen H,et al.Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors[J].J Biol Chem,2008,283(52):36234-36240.
[11] Fang JY,Richardson BC.The MAPK signalling pathways and colorectal cancer[J].LancetOncol,2005,6(5):322-327.
[12]Sasaki K,Hitora T,Nakamura O,et al.The role of MAPK pathway in bone and soft tissue tumors[J].Anticancer Res,2011,31(2):549-553.
[13] Lin H,Li Z,Chen C,et al.Transcriptional and post-transcriptional mechanisms for oncogenic overexpression of etherà go-go K+channel[J].PLoSOne,2011,6(5):e20362.
[2015-06-03收稿][2015-07-15修回][编辑江德吉]
The p38MAPK/p53 pathway regulatesexpression of Etheràgo-go in osteosarcoma
Wu Jin1,Liu Qingjun1,Chen Zhida1,Zeng Wenrong1,Wu Xinyu2,Lin Bin1(1Department of Orthopaedics,2Department of Neurology,The Affiliated Southeast Hospital of Xiamen University,Zhangzhou 363000,P.R.China)
Lin Bin.E-mail:linbin813@163.com
Objective To detect the expression of Etheràgo-go(Eag)in human osteosarcoma and examine possible pathways regulating Eag expression.Methods Eag expression was analyzed in the osteosarcoma cell line MG-63 using reverse transcription-polymerase chain reaction and western blotting.Effects of Eag inhibition on cell proliferation were examined in MG-63 cultures,and effects of short hairpin RNA-mediated knockdown of Eag on osteosarcoma growth were examined in an in vivo xenograft model.Activation of the mitogen-activated protein kinase(MAPK)/p53 signaling pathway in MG-63 cells was detected using Western blot analysis.Results Eag is overexpressed in MG-63 cells,and imipramine or Eag short hairpin RNA significantly inhibited MG-63 proliferation in vitro and in vivo.MG-63 proliferation was also strongly inhibited by the p38 MAPK inhibitor SB203580 or small interfering RNA(siRNA).Using SB203580 or siRNA to inhibit p38 MAPK activation reduced levels of Eag protein but increased levels of p53 protein.Using nutlin-3 to activate p53 reduced levels of Eag protein and arrested MG-63 growth,while using pifithrin-alpha to inactivate p53 increased the levels of Eag and promoted MG-63 growth.Conclusion The Eag gene functions as an oncogene to promote the proliferation of osteosarcoma cells,and the p38 MAPK/p53 pathway regulates high Eag expression in osteosarcoma cells.
Bone neoplasm;Etheràgo-go;Cell proliferation;MAPK pathway;p53 gene;Regulation;Expression
R738.1
A
1674-5671(2015)05-05
10.3969/j.issn.1674-5671.2015.05.03
国家自然科学基金资助项目(81402217)
林斌。E-mail:linbin813@163.com
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
理财周刊(2022年4期)2022-04-30
昆明医科大学学报(2022年1期)2022-02-28
天津医科大学学报(2021年4期)2021-08-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
现代临床医学(2021年2期)2021-03-29
陕西医学杂志(2020年10期)2020-12-27
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
听力学及言语疾病杂志(2019年1期)2019-01-14
