表面钒修饰对α-Fe2O3材料光电化学性能的增强作用
2015-11-03 08:53:05姚利珍孔德生杜玖瑶张经纬李文娟冯媛媛
物理化学学报 2015年10期
姚利珍 孔德生杜玖瑶 王 泽 张经纬 王 娜 李文娟 冯媛媛
(曲阜师范大学化学与化工学院,山东 曲阜 273165)
表面钒修饰对α-Fe2O3材料光电化学性能的增强作用
姚利珍 孔德生*杜玖瑶 王 泽 张经纬 王 娜 李文娟 冯媛媛
(曲阜师范大学化学与化工学院,山东 曲阜 273165)
对半导体材料进行表面化学修饰或改性, 是提高其光催化活性、有效利用光能的一种重要措施. 本文结合水热化学法、化学池沉积和后续热处理等, 分别制备了未修饰α-Fe2O3和钒修饰的α-Fe2O3光电极材料. 利用X射线粉末衍射(XRD)谱和紫外-可见漫反射光谱(UV-Vis-DRS)技术分析表征了材料的晶相结构、化学组成和光谱吸收等固体物理化学性能; 利用光电流测量和电化学交流阻抗谱(EIS)实验技术, 并基于1 molL-1NaOH (pH 13.6)中的光电化学水分解反应, 研究了钒修饰对α-Fe2O3材料光电化学性能的增强作用. 结果表明,与未修饰的Fe2O3材料相比, 钒修饰α-Fe2O3样品出现FeVO4的XRD特征峰, 但临界光吸收波长未发生红移; 钒修饰使Fe2O3材料的光电流增大4-5倍、光生载流子在电极表面的复合几率降低了3/4-4/5、电极表面电荷传递速率(表观一级速率常数)明显提高. 结合Fe2O3/溶液界面半导体能带模型和有关研究结果, 分析了研究体系的界面电荷动力学传输过程以及钒修饰使α-Fe2O3材料光电化学性能增强的原因.
水分解; 光生电荷传输; 表面复合; 光电流; 交流阻抗谱
1 引 言
氢气具有高储能无污染的特点,被认为是传统化石能源的最佳替代品. 利用太阳能实现大规模光电化学(PEC)分解水制氢(将太阳能转化为化学能),是国内外化学与材料研究者共同努力的一个目标.1,2半导体光电极是PEC分解水制氢系统中的核心构件. 金属氧化物半导体材料(如TiO2、Fe2O3、WO3、ZnO2、SrTiO4、BiVO4等)具有稳定性高、价格低廉、绿色环保、易于制备等特点,是目前光电化学研究中备受关注的光电极基本材料.3-5
与其它常用的n型半导体氧化物材料相比,α-Fe2O3具有较适宜的禁带宽度(Eg= 2-2.2 eV),可吸收波长小于550-600 nm的太阳光(光子吸收近40%),AM 1.5光照条件下的理论光电转化效率可达12.9%,被视为最具实际应用前景的光电极材料之一.3-6但由于α-Fe2O3自身具有较低的电子迁移率(10-2-10-1cm2V-1s-1)、较短的空穴扩散长度(2-4 nm)、较慢的表面析氧速率等不足,致使光生载流子本体复合及表面复合严重,实际光电转化效率远低于理论值,严重制约着α-Fe2O3作为光电极材料的实际应用.
为提高α-Fe2O3材料的PEC活性,近几年来人们在以下三个方面开展了大量研究工作: 一是制备纳米结构的α-Fe2O3材料,如纳米管、纳米棒、纳米颗粒等α-Fe2O3材料;1,7-9二是利用其它元素对α-Fe2O3进行本体掺杂,常见的掺杂离子(或原子)主要有Nb5+、V5+、Ti4+、Zr4+、Al3+、Cr3+、Co3+、Ir3+、C、Si、S、N、P、F等;1,9-14三是利用其它元素或半导体材料对α-Fe2O3进行表面修饰,已报道的表面修饰材料包括Ag、Pt、C、氧化钴(CoOx)、氧化铱(IrOx)、磷酸盐等.15-21有关研究结果表明,对α-Fe2O3进行本体掺杂和表面修饰是增强α-Fe2O3电极PEC活性的两种有效途径,但二者的作用机制不同. 在本体掺杂过程中,局外离子或原子将作为替位或填隙离子嵌入Fe2O3晶格,从而使掺杂α-Fe2O3材料的本体电子性能或光吸收性能得到改善,如(i) 提高了掺杂Fe2O3材料的本体电荷密度及电导率,使光生载流子本体复合受到抑制;5,22,23(ii) 禁带宽度减小,增强了材料的可见光吸收活性.13,24而表面修饰则对α-Fe2O3材料的表面物理化学性能产生影响,不同表面修饰材料对增强α-Fe2O3电极PEC活性的可能原因主要有以下几个方面: (i) 电极的光谱吸收临界波长发生红移,进一步增强了可见光吸收活性;15(ii) 存在表面等离子体共振(SPR)增强效应,促进了可见光吸收;25,26(iii) 促进了光生电子-空穴对的分离,表面复合过程受到抑制;20,21(iv) 催化了光电极表面的水氧化反应,提高了表面/界面电荷传递速率等.2,19
由于α-Fe2O3具有较好的可见光吸收性能,该氧化物半导体材料不仅可用于太阳能分解水制氢(如上所述),而且可用于光催化降解环境有机污染物,8,12,27或作为光吸收层用于对宽带隙半导体材料(如TiO2)进行表面修饰.28-30因此,研究高活性α-Fe2O3光电极材料的制备与性能,将非常有助于促进α-Fe2O3材料在太阳能利用领域中的实际应用. 同时,虽然有人研究过钒离子对α-Fe2O3光电极材料的本体掺杂作用以及Fe2O3-FeVO4二元体系的光催化或化学催化作用,31-35但到目前为止,文献中尚未见有关钒对α-Fe2O3表面修饰作用的光电化学研究报道.近期我们对TiO2纳米管、Fe2O3颗粒、棒状Ca3Bi8O15等半导体材料的制备、光电化学分解水制氢、光催化降解无机污染物、有关反应机理等方面,开展了部分研究工作,36-42本文中我们利用水热化学法分别制备了α-Fe2O3和钒修饰α-Fe2O3光电极材料,并基于PEC水分解反应体系研究了钒修饰对α-Fe2O3光电化学活性的增强作用及原因.
2 实验部分
2.1 钒修饰α-Fe2O3光电极材料的制备
首先利用溶胶-凝胶法制备Fe(OH)3胶体,将10 mL FeCl3饱和溶液在搅拌条件下滴加至沸腾的40 mL蒸馏水中,保持沸腾一定时间以使过多的水份蒸发,在半透膜中渗析-纯化,得到棕红色Fe(OH)3溶胶;然后采用刮涂法将Fe(OH)3胶体均匀地涂覆在钛片表面上,控制膜厚100 μm、面积1 cm2,并在马弗炉中于550 °C和大气条件下热处理1.5 h,制得α-Fe2O3光电极,本文标记为“Fe2O3”材料.
将制得的Fe2O3电极分别放入25或90 °C 的0.1 molL-1VOSO4溶液中浸渍1 h,然后在马弗炉中于550 °C和大气条件下热处理1.5 h,得到钒修饰Fe2O3电极,本文分别标记为“V@Fe2O325 °C”、“V@Fe2O390 °C”材料. 为表征电极材料的晶相结构和光学性能,分别在相应条件下制备了Fe2O3、V@Fe2O325 °C、V@Fe2O390 °C粉体材料. 所用溶液均由分析级试剂和利用石英亚沸蒸馏器制得的二次蒸馏水配制. 光电极钛片基底(纯度99.7%,Sigma-Aldrich)宽1 cm、长6 cm、厚度0.127 mm,使用前进行表面打磨,并在0.1 molL-1HClO4+ 0.02 molL-1NaF溶液中浸泡清洗30 s.
2.2 性能表征
光电化学实验装置为H形双室三电极体系. 电解池带有直径3.5 cm的石英玻璃窗口,以使入射光垂直照射光电极表面,以所制备光电极为光阳极(电极表面Fe2O3或V@Fe2O3光活性层的面积1 cm2)、铂片为对电极、饱和甘汞电极(SCE)为参比电极,电解质溶液为1 molL-1NaOH溶液(pH 13.6). 实验仪器包括CHI600d型电化学工作站(上海辰华仪器公司)、AM 1.5太阳光模拟器(LSXS500,Zolix卓立汉光),入射光强度100 mWcm-2. 光电流-电位(Iph-E)曲线测量时的电位扫描速率为5 mVs-1,光电流-时间(Iph-t)曲线测量时光照开/关转换频率为0.05 Hz,电化学交流阻抗谱(EIS) 测量频率为1-105Hz(幅值10 mV). Iph-E曲线和EIS谱分别在恒电位0.3 V(vs SCE) (即0.54 V (vs NHE(标准氢电极)))下测量.
X射线衍射(XRD)谱利用Miniflex 600型XRD粉末衍射仪测量(Rigaku Co. Ltd.,Japan),Cu Kα辐射(波长0.15406 nm),扫描速率为4°(2θ)min-1,紫外-可见漫反射光谱(UV-Vis-DRS)利用UV-3600型紫外/可见/近红外分光光度计测量(SHIMADZU Co.,Japan),积分球 + BaSO4背底,扫描速率为200 nmmin-1. EIS、XRD和UV-Vis-DRS测量结果分别利用ZSimpWin程序、Jade软件包和Kubelka-Munk函数分析处理.43-45
3 结果与讨论
3.1 晶相结构与光吸收性能
利用XRD粉末衍射技术表征了所制备Fe2O3、以及分别在25和90 °C下所制备的钒修饰Fe2O3样品的晶相结构,如图1所示. 由图1(a) 可见,经过550 °C热处理后的Fe2O3为α-Fe2O3(hematite)晶型,其特征衍射峰(104)、(110)、(024)、(116)等与标准卡JCPDS 33-0644 (图1(d))较好地吻合,特征衍射峰峰形尖锐,说明材料的结晶程度较高. α-Fe2O3样品的平均晶粒度D可利用最强衍射峰(104)并根据Scherrer方程确定:46

式中λ为 X 光的入射波长,β 为衍射峰的半峰宽(FWHM),θ为衍射角. 利用Jade软件所确定的(104)晶面垂直方向上的平均晶粒度约为 77 nm.
钒修饰使Fe2O3材料的XRD谱峰数量明显增多(图1(b,c)). 分析表明,在室温25 °C下所制备钒修饰Fe2O3样品的XRD谱中(图1(b))出现了FeVO4的(012)、(130)、(311)、 (004)、(400)特征峰,且与标准卡JCPDS 89-0618 (图1(d))较好地吻合,说明所形成的FeVO4为正交晶格结构; 而在90 °C下所制备钒修饰Fe2O3样品的XRD谱中(图1(c))则进一步出现了FeVO4的(112)和(040)谱峰. 含钒物种FeVO4的存在,证明了本文钒修饰Fe2O3材料制备方法的有效性;XRD结果同时表明,在较高温度(90 °C)时所制备钒修饰Fe2O3材料中的FeVO4晶相结构更加完整和典型,该结果与材料在光照条件下所表现出的不同光电化学行为或性能较好地对应.
为探究钒修饰使α-Fe2O3材料光电化学活性增强的可能原因,进一步研究了三种光电极材料的光学性能(如图2所示). 图2(a)是测量的可见光区的UV-Vis-DRS谱,图2b是根据Kubelka-Munk函数(式(2)) 和Tauc方程(式(3))得到的 [F(R)E]2-E曲线.

式中F(R)为Kubelka-Munk函数;44,45R为漫反射系数;h为Plank常数; v为入射光频率,hv为光子能量(eV);A为常数; Eg为半导体禁带宽度; n为能带跃迁指数,n = 2为直接跃迁,n = 1/2为间接跃迁. 由图2(b)可以看出,钒修饰并未引起Fe2O3材料禁带宽度的变化,三种光电极材料的禁带宽度基本相同(Eg≈ 2.05 eV),且三种Fe2O3材料均为直接跃迁. 该结果表明,钒对α-Fe2O3的表面修饰对材料本体光吸收性能的影响很小,不会引起材料临界光吸收波长的红移.
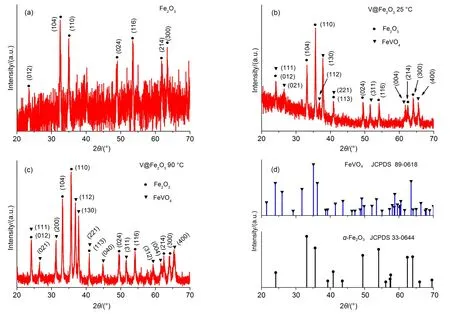
图1 光电极材料的XRD图谱Fig.1 XRD patterns of the photoelectrode materials

图2 Fe2O3及钒修饰Fe2O3光电极材料的反射谱(a)与Tauc图(b)Fig.2 Reflection spectra (a) and Tauc Fe2O3(b) of Fe2O3and V-modified Fe2O3samples
3.2 光电流响应
通过光电流测量研究了钒的表面修饰对Fe2O3材料光电化学性能的影响. 图2是利用三电极体系在1 molL-1NaOH (pH 13.6)溶液中测量的Iph-E曲线. 本研究所用电解质溶液为NaOH,不含其它电活性物种,有关电极过程对应于水的光电化学分解:

由图2可见,与未修饰的Fe2O3电极相比,钒修饰Fe2O3电极的光电流明显增大,且在90 °C下钒修饰Fe2O3样品的光电化学活性高于在室温25 °C下钒修饰的Fe2O3样品. 另外,三种电极在无光照条件下的暗电流很小,在E < 0.5 V (vs SCE)电位范围内的暗电流密度为10-7-10-5Acm-2,故可将光照条件下的电流值视为光电流(Iph).
图3为恒电位0.3 V (vs NHE)下的暂态Iph-t转换曲线. 由图3可见,在光照瞬间光电流密度急剧上升并达到峰值Iph(max) (见图中标注),然后逐渐降低达到稳态. 经过3次光照开/关循环后,光电流密度相应逐渐趋于稳定,三种电极材料在140 s时的峰值光电流密度Iph(max)和160 s时的暂态光电流密度Iph(ss)见表1. 由表1可见,与未修饰的Fe2O3电极相比,在室温(25 °C)的含钒溶液中所制备的钒修饰Fe2O3电极V@Fe2O325 °C的Iph(ss) = 0.096 mAcm-2,约为未修饰Fe2O3的3倍; 在90 °C的含钒溶液中所制备的V@Fe2O390 °C电极的Iph(ss) = 0.14 mAcm-2,约为未修饰Fe2O3的4倍. 这表明,钒修饰使Fe2O3材料的光电化学活性显著增强,且在较高温度下(90 °C)所制备钒修饰Fe2O3材料的光电化学活性更高,该结果与Iph-E曲线测量结果(图2)相吻合.

图3 Fe2O3及钒修饰Fe2O3光电极的光电流密度-电位曲线Fig.3 Iph-E curves of Fe2O3and V-modified Fe2O3photoelectrodes
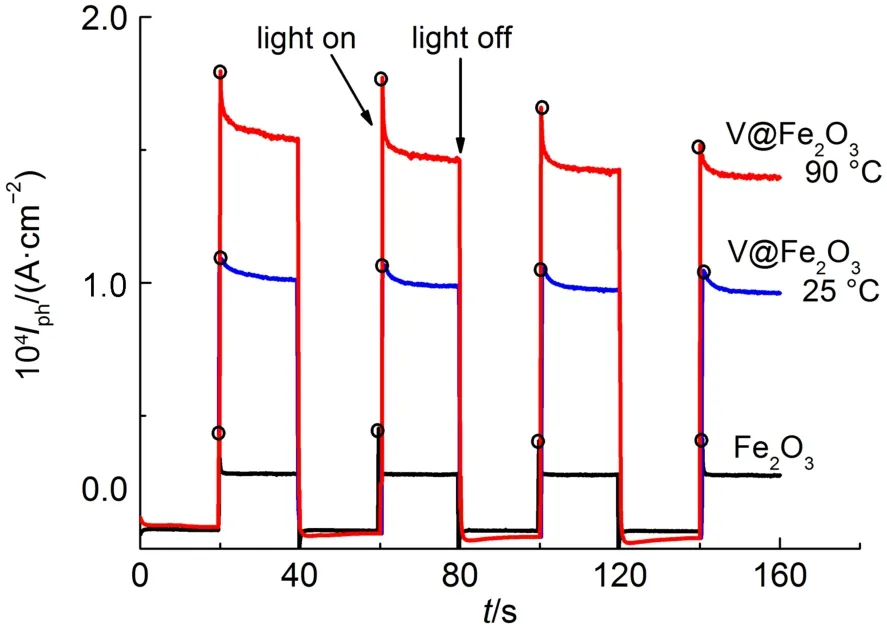
图4 Fe2O3及钒修饰Fe2O3光电极的暂态光电流密度响应Fig.4 Transient photocurrent density responses of Fe2O3and V-modified Fe2O3photoelectrodes

表1 Fe2O3与钒修饰Fe2O3光电极的峰值光电流密度(Iph(max)),暂态光电流密度(Iph(ss))及载流子表面复合几率Table 1 Values of peak photocurrent densities (Iph(max)) and transient photocurrent densities (Iph(ss)),and apparent surface-recombination probability of charge-carriers for Fe2O3and V-modified Fe2O3photoelectrodes
光照开始后光电流密度由Iph(max)快速衰减为Iph(ss) (图3),其原因主要是由于光生电子-空穴对在“光电极/溶液”界面发生的表面复合过程所致.36,47,48因此,钒修饰对光电极Iph-t曲线(图4)暂态转换行为的影响,反映了钒修饰对光生载流子在Fe2O3电极表面复合过程的影响,并可用表观表面复合几率(SPR)表征之:36
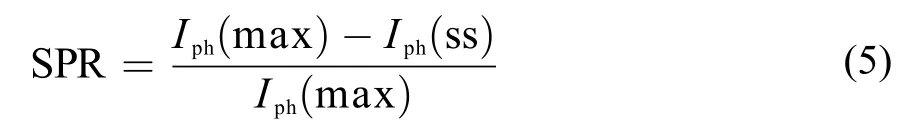
根据式(5)和表1中的Iph(ss)及Iph(max)数据所确定的三种电极材料的表观复合几率列于表1中. 可以看出,Fe2O3材料表面经过钒修饰后,表观SPR仅为未修饰Fe2O3材料的1/4-1/5,光生载流子的表面复合过程受到明显的抑制. 由于光生导带(CB)电子与价带(VB)空穴之间的表面复合是对已吸收光子的直接损耗,因此该结果表明,与未修饰的Fe2O3电极相比,钒修饰Fe2O3电极对所吸收光子的有效利用率(即光电转化效率)提高了约75%-80%. 为进一步探究钒修饰对Fe2O3材料光电化学性能的增强作用(以及对光生载流子表面复合过程的抑制作用)的原因,本文利用交流阻抗技术研究了光照条件下电极/溶液界面的电荷传输动力学性能.
3.3 交流阻抗谱测量
交流阻抗技术是研究各种电极/溶液界面电荷传输动力学性能的重要手段.39,49图5为Fe2O3、V@Fe2O325 °C、V@Fe2O390 °C三种光电极在1 molL-1NaO溶液中、光照条件下测量的EIS谱. 由图5可见,三种光电极材料的界面阻抗响应均表现为2个时间常数,这与文献报道结果11,50,51类似. 结合文献中有关金属氧化物半导体材料(特别是TiO2和Fe2O3)表面PEC析氧反应机理的阻抗研究,18,36,37,38,50,52高频时间常数可归因于光生空穴在“光电极/溶液”界面的Faraday电荷传输过程,低频时间常数可归因于阳极析氧过程中电极表面所形成的中间含氧物种(如羟基自由基―OH*或过氧化物―O―O―)的吸/脱附过程. 图5同时给出了相应的等效电路,其CDC描述码为 Rs(Cdl(Rct(RaQ))),其中Rs为溶液电阻,Cdl为电极/溶液界面Helmholtz双层电容,Rct为界面电荷传输电阻,Ra是与中间物种吸脱附过程有关的等效电阻,Q为常相位角元件(CPE),CPE的阻抗为ZCPE= Y0-1(jω)-n,27,49其中Y0和n分别是与频率无关的CPE参数.

图5 Fe2O3及钒修饰Fe2O3光电极的EIS 图Fig.5 EIS Nyquist plots of Fe2O3and V-modified Fe2O3photoelectrodes

图6 测量与计算的Fe2O3和钒修饰Fe2O3光电极的Bode频谱图Fig.6 Measured and calculated Bode plots of Fe2O3and V-modified Fe2O3photoelectrodes
图6为利用等效电路模型(图5)得到的拟合频谱与测量频谱的Bode图,可见二者基本吻合,说明所使用的等效电路模型能较好地描述电极表面的光电化学过程,计算确定的有关等效电路元件参数见表2. 由表2可见,钒修饰Fe2O3电极的Rct和Ra值均明显小于未修饰Fe2O3电极的Rct和Ra值,反映出钒修饰对界面电荷传输过程和光生中间物种吸脱附过程的改善作用. 电极/溶液界面Faraday电荷传输过程动力学是决定电极光电流及其大小的内在因素,该过程的表观准一级速率常数k可由下式确定:

根据式(6)及CdlRct数据,三种光电极在1 molL-1NaOH (pH 13.6)溶液中的界面电荷传输过程的准一级速率常数分别为k(Fe2O3) = 1302.9 s-1、k(V@Fe2O325 °C) = 1467.1 s-1、k(V@Fe2O390 °C) = 2966.2 s-1(表2). 该结果与分别利用三种电极所测量到的光电流大小(图3、图4)较好地吻合. 因此,钒修饰对Fe2O3材料光电化学活性的增强作用,可归因于钒修饰对“光电极/溶液”界面处光生电荷界面传输动力学过程及性能的改善. 同时,在较高温度(90 °C)下所制备的钒修饰Fe2O3电极界面电荷传输速率高于在较低温度(25°C)下所制备的钒修饰Fe2O3电极,其原因可能是较高的温度更有利于钒离子物种与Fe2O3表面发生物理化学相互作用,从而使材料表面修饰的FeVO4晶相结构更为完整(图1(c)),对阳极析氧反应的光电催化活性更高.
界面电荷传递速率常数数值的大小与光照条件、电极材料、施加电位、测量方法及理论模型等因素有关.11,38,50最近,Bertoluzzi和Bisquert52针对“半导体/溶液”界面的PEC水分解反应,提出了一个一般化的EIS动力学物理模型,不同情况下半导体光电极表面电荷传输过程的一级速率常数(ks)的典型值为101-103s-1数量级(参见文献52中的图3和图5). Peter与其合作者20,50分别利用EIS和强度调制光电流谱(IMPS)技术,研究了Fe2O3和钴修饰Fe2O3光阳极表面析氧动力学(入射光波长λ = 455 nm,1 molL-1NaOH溶液中),所确定的电荷传输过程的一级速率常数(kr)的数值为100-102s-1. 这些结果与本文所确定的一级速率常数(k)的数值相近(虽然不同作者所使用的理论模型不同).

表2 光电极材料的EIS 等效电路元件参数Table 2 Parameters of EIS equivalent electrical elements for the photoelectrode materials

图7 研究体系的能带模型及“光电极/溶液”界面电荷传输过程示意图Fig.7 Schematic depiction of band energy level and interfacial charge transfer for the “photoelectrode/solution”interface under investigation
3.4 “光电极/溶液”界面能级模型

另一方面,一部分空穴将陷入能级位于禁带中的表面态,11,36,51并通过表面态与导带电子发生表面复合(即过程①,图7(b)):

过程①与过程②相互竞争,共同捕获价带空穴.20,36光生空穴向溶液一侧的传递速率越快,则表面复合几率越小,通过外电路检测到的光电流响应越大. 因此可以认为,由于钒对Fe2O3的表面修饰改善了电极/溶液界面电荷传输性能,钒修饰电极“V@Fe2O390 °C”的准一级速率常数是Fe2O3电极准一级速率常数的2.28倍(表2),从而使光生电荷在电极表面的复合几率SPR显著降低(表1),电极的光电流密度明显增大(图3、图4),这是钒修饰使Fe2O3材料光电化学活性增强的原因.
另外值得注意的是,矾酸盐(包括FeVO4)是一类新型的n型半导体光催化剂,FeVO4禁带宽度Eg为2-2.2 eV (与Fe2O3相近),53-55具有可见光活性. 近期研究表明,FeVO4或FeVO4-Fe2O3二元体系不仅在光照下可用于环境有机污染物的氧化降解,33,55-57而且在无光照条件下对有机物 (如亚甲基蓝、苯、甲苯、醇类分子等)的化学氧化反应具有显著的催化(FeVO4)与协同催化(FeVO4-Fe2O3)作用.31,34,58-60这些结果与本文所观察到的钒修饰对Fe2O3表面水的PEC氧化分解反应的促进作用具有一定的相似性和一致性.
4 结 论
利用XRD、UV-Vis-DRS、光电流测量和交流阻抗实验技术,研究了钒修饰对α-Fe2O3光电极材料光电化学性能的增强作用及原因. 基于本文实验结果得到以下结论:
(1) 结合水热化学、室温(25 °C)和90 °C VOSO4溶液化学池沉积(25和90 °C VOSO4溶液中)和后续550 °C热处理等方法,分别制备了未修饰Fe2O3和2种钒修饰Fe2O3光电极材料; XRD和UV-Vis-DRS结果表明,钒修饰α-Fe2O3样品表面存在正交结构的FeVO4晶相,但钒修饰对材料的可见光吸收性能无明显影响;
(3) 利用交流阻抗谱测量研究了“光电极/溶液”界面处的Faraday电荷传输性能,结果表明,表面钒修饰对α-Fe2O3材料光电化学性能的增强作用以及对光生电荷表面复合过程的抑制作用,可归因于钒修饰对“光电极/溶液”界面电荷传输性能的改善与增强作用,钒修饰电极“V@Fe2O390 °C” 和未修饰Fe2O3电极的界面电荷传输准一级速率常数分别为k(V@Fe2O390 °C) = 2966.2 s-1、k(Fe2O3) = 1302.9 s-1;结合Fe2O3/溶液界面能带模型,分析了Faraday电荷传输与表面复合过程对研究体系光电化学相应的影响.
(1)Yang,X.; Liu,R.; He,Y.; Thorne,J.; Zheng,Z.; Wang,D. Nano Res. 2015,8,56. doi: 10.1007/s12274-014-0645-2
(2)van de Krol,R.; Grätzel,M. Introduction,Principles of Photoelectrochemical Cells. In Photoelectrochemical Hydrogen Production; van de Krol,R.,Grätzel,M. Eds.; Springer Science + Business Media: New York,2012; pp 3-67.
(3)Valdés,A.; Brillet,J.; Grätzel,M.; Gudmundsdóttir,H.; Hansen,H. A.; Jónsson,H.; Klüpfel,P.; Kroes,G. J.; Le Formal,F.; Man,I. C.; Martins,R. S.; Nørskov,J. K.; Rossmeisl,J.; Sivula,K.;Vojvodic,A.; Zäch,M. Phys. Chem. Chem. Phys. 2012,14,49. doi: 10.1039/C1CP23212F
(4)Chen,X.; Shen,S.; Guo,L.; Mao,S. S. Chem. Rev. 2010,110,6503. doi: 10.1021/cr1001645
(5)Zhou,W.; Xie Q.; Lian,S. Prog. Chem. 2013,25,1989. [周文理,谢青季,廉世勋. 化学进展,2013,25,1989.]
(6)Diab,M.; Mokari,T. Inorg. Chem. 2014,53. 2304.
(7)Rangaraju,R. R.; Raja,K. S.; Panday,A.; Misra,M. Electrochim. Acta 2010,55,785. doi: 10.1016/j.electacta.2009.07.012
(8)Pradhan,G. K.; Padhi,D. K.; Parida,K. M. ACS Appl. Mater. Interfaces 2013,5,9101. doi: 10.1021/am402487h
(9)Lee,C. Y.; Wang,L.; Kado,Y.; Kirchgeorg,R.; Schmuki,P. Electrochem. Commun. 2013,34,308. doi: 10.1016/j.elecom.2013.07.024
(10)Wang,L.; Lee,C.Y.; Schmuki,P. Electrochem. Commun. 2013,30,21. doi: 10.1016/j.elecom.2013.01.013
(11)Shangguan,P. P.; Tong,S. P.; Li,H. L.; Leng,W. H. Acta Phys. -Chim. Sin. 2013,29,1954. [上官鹏鹏,童少平,李海丽,冷文华. 物理化学学报,2013,29,1954.] doi: 10.3866/PKU.WHXB201306261
(12)Jin,H.; Wang,J.; Ji,Y.; Chen,M. M.; Zhang,Y.; Wang,C.;Cong,Y. Q. Acta Phys.-Chim. Sin. 2015,31,955. [金 环,王 娟,姬 云,陈媚媚,张 轶,王 齐,丛燕青. 物理化学学报,2015,31,955.] doi: 10.3866/PKU.WHXB201503112
(13)Kumar,P.; Sharma,P.; Shrivastav,R.; Dass,S.; Satsangi,V. R. Int. J. Hydrog. Energy 2011,36,2777. doi: 10.1016/j.ijhydene.2010.11.107
(14)Zhang,X.; Li,H.; Wang,S.; Fan,F. R. F.; Bard,A. J. J. Phys. Chem. C 2014,118,16842. doi: 10.1021/jp500395a
(15)Shaban,Y. A.; Khan,S. U. M. Sci. Adv. Mater. 2012,4,356. doi: 10.1166/sam.2012.1292
(16)Barroso,M.; Mesa,C. A.; Pendlebury,S. R.; Cowan,A. J.;Hisatomi,T.; Sivula,K.; Grätzel,M.; Klug,D. R.; Durrant,J. R. PNAS 2012,109,15640. doi: 10.1073/pnas.1118326109
(17)Barroso,M.; Cowan,A. J.; Pendlebury,S. R.; Grätzel,M.; Klug,D. R.; Durrant,J. R. J. Am. Chem. Soc. 2011,133,14868. doi: 10.1021/ja205325v
(18)Klahr,B.; Gimenez,S.; Fabregat-Santiago,F.; Bisquert,J.;Hamann,T. W. J. Am. Chem. Soc. 2012,134,16693. doi: 10.1021/ja306427f
(19)Shen,S.; Zhou,J.; Dong,C. L.; Hu,Y.; Tseng,E. N.; Guo,P.;Guo,L.; Mao,S. S. Sci. Rep. 2014,4,6627. doi: 10.1038/srep06627
(20)Peter,L. M.; Wijayantha,K. G. U.; Tahir,A. A. Faraday Discuss. 2012,155,309. doi: 10.1039/C1FD00079A
(21)Sun,W.; Meng,Q.; Jing,L.; Liu,D.; Cao,Y. J. Phys. Chem. C 2013,117,1358. doi: 10.1021/jp309599d
(22)Augustynski,J.; Alexander,B. D.; Solarska,R. Top. Curr. Chem. 2011,303,1. doi: 10.1007/978-3-642-22294-8
(23)Varshney,D.; Yogi,A. J. Mol. Struct. 2011,995,157. doi: 10.1016/j.molstruc.2011.04.011
(24)Martis,V.; Oldman,R.; Anderson,R.; Fowles,M.; Hyde,T.;Smith,R.; Nikitenko,S.; Bras,W.; Sankar,G. Phys. Chem. Chem. Phys. 2013,15,168. doi: 10.1039/C2CP43307A
(25)Sun,L.; Wu,W.; Yang,S.; Zhou,J.; Hong,M.; Xiao,X.; Ren,F.; Jiang,C. ACS Appl. Mater. Interfaces 2014,6,1113. doi: 10.1021/am404700h
(26)Zhou,W.; Li,T.; Wang,J.; Qu,Y.; Pan,K.; Xie,Y.; Tian,G.;Wang,L.; Ren,Z.; Jiang,B.; Fu,H. Nano Res. 2014,7,731. doi: 10.1007/s12274-014-0434-y
(27)Jana,S.; Mondal,A. ACS Appl. Mater. Interfaces 2014,6,15832. doi: 10.1021/am5030879
(28)Huang,Y. C.; Zhao,Z. F.; Li,S. X.; D,J.; Zheng,H. J. Chin. J. Inorg. Chem. 2015,31,133. [黄益操,赵浙菲,李世雄,邸 婧,郑华均. 无机化学学报,2015,31,133.]
(29)Xu,Z.; Huang,C.; Wang,L.; Pan,X.; Qin,L.; Guo,X.; Zhang,G. Ind. Eng. Chem. Res. 2015,54,4593. doi: 10.1021/acs.iecr.5b00335
(30)Mohapatra,S. K.; Banerjee,S.; Misra,M. Nanotechnology 2008,19,315601. doi: 10.1088/0957-4484/19/31/315601
(31)Oliveira,H. S.; Oliveira,L. C. A.; Pereira,M. C.; Ardisson,J. D.;Souza,P. P.; Patricio,P. O.; Moura,F. C. C. New J. Chem. 2015,39,3051. doi: 10.1039/C4NJ02063D
(32)Hwang,H. K.; Seo,J. W.; Seo,W. S.; Lim,Y. S.; Park,K. Int. J. Energy Res. 2014,38,241. doi: 10.1002/er.v38.2
(33)Wang,M.; Wang,L. A.; Zhou,L. N.; Zhang,W. J. J. Chin. Ceram. Soc. 2009,37,203. [王 敏,王里奥,周丽娜,张文杰.硅酸盐学报,2009,37,203.]
(34)Zhang,G. Q.; Zhang,X.; Lin,T.; Gong,T.; Qi,M. Chin. Chem. Lett. 2012,23,145. doi: 10.1016/j.cclet.2011.10.015
(35)Zhang,G. Q.; Zhang,X.; Qi,M.; Lin,T.; Gong,T. Chin. J. Catal. 2012,33,870. [张贵泉,张 昕,祁 敏,林 涛,龚 婷. 催化学报,2012,33,870.]
(36)Kong,D. S.; Wei,Y. J.; Li,X. X.; Zhang,Y.; Feng,Y. Y.; Li,W. J. J. Electrochem. Soc. 2014,161,H144.
(37)Kong,D. S.; Zhang,X. D.; Wang,J.; Wang,C.; Zhao,X.; Feng,Y. Y.; Li,W. J. J. Solid State Electrochem. 2013,17,69. doi: 10.1007/s10008-012-1854-9
(38)Zhang,J. W.; Kong,D. S.; Zhang,H.; Du,D. D.; Wang,N.;Feng,Y. Y.; Li,W. J. J. Solid State Electrochem. 2015,accepted. doi: 10.1007/s10008-015-2948-y
(39)Kong,D. S. Langmuir 2010,26,4880. doi: 10.1021/la9036869
(40)Li,W. J.; Du,D. D.; Yan,T. J.; Kong,D. S.; You,J. M.; Li,D. Z. J. Colloid Interface Sci. 2015,444,42. doi: 10.1016/j.jcis.2014.12.052
(41)Du,D. D; Li,W. J.; Chen,S. S.; Yan,T. J.; You,J. M.; Kong,D. S. New J. Chem. 2015,39,3129.
(42)Li,W. J.; Kong,D. S.; Cui,X. L.; Du,D. D.; Yan,T. J.; You,J. M. Mater. Res. Bull. 2014,51,69. doi: 10.1016/j.materresbull.2013.12.007
(43)ZSimpWim Echem Software,Version 3.20d,2004; Jade 5.0(MDI) software package,2004.
(44)Barron,V.; Torrent,J. J. Soil Sci. 1986,37,499. doi: 10.1111/ejs.1986.37.issue-4
(45)Li,W.; Li,D.; Chen,Z.; Huang,H.; Sun,M.; He,Y.; Fu,X. J. Phys. Chem. C 2008,112,14943. doi: 10.1021/jp8049075
(46)Boudjemaa,A.; Boumaza,S.; Trari,M.; Bouarab,R.; Bouguelia,A. Int. J. Hydrog. Energy 2009,34,4268. doi: 10.1016/j.ijhydene.2009.03.044
(47)de Tacconi,N. R.; Boyles,C. A.; Rajeshwar,K. Langmuir 2000,16,5665. doi: 10.1021/la000037x
(48)Radecka,M.; Wierzbicka,M.; Komornicki,S.; Rekas,M. Phys. B 2004,348,160. doi: 10.1016/j.physb.2003.11.086
(49)Bonanos,N.; Steele,B. C. H.; Butler,E. P.; MacDonald,J. R.;Johnson,W. B.; Worrell,W. L.; MacDonald,D. D.; McKubre,M. C. H.; Barsoukov,E.; Conway,B. E.; Wagner,N. Applications of Impedance Spectroscopy. In Impedance Spectroscopy: Theory,Experiment,and Applications,2nd ed.;Barsoukov,E.,MacDonald,J. R. Eds.; John Wiley & Sons,Inc.: New Jersey,2005; pp 205-537.
(50)Wijayantha,K. G. U.; Saremi-Yarahmadi,S.; Peter,L. M. Phys. Chem. Chem. Phys. 2011,13,5264. doi: 10.1039/c0cp02408b
(51)Klahr,B.; Gimenez,S.; Fabregat-Santiago,F.; Hamann,T.;Bisquertm,J. J. Am. Chem. Soc. 2012,134,4294. doi: 10.1021/ja210755h
(52)Bertoluzzi,L.; Bisquert,J. J. Phys. Chem. Lett. 2012,3,2517. doi: 10.1021/jz3010909
(53)Rao,N. S.; Palanna,O. G. Bull. Mater. Sci. 1995,18,229. doi: 10.1007/BF02749660
(54)Li,A. T.; Cao,L. Y.; Huang,J. F.; Huang,Y. C.; Wu,J. P. J. Synth. Cryst. 2012,41,1227. [李阿婷,曹丽云,黄剑锋,黄毅成,吴建鹏. 人工晶体学报,2012,41,1227.]
(55)Wang,M.; Luan,H. Y.; Yu,P.; Che,Y. S.; Niu,C.; Dong,D. Chin. J. Nonferrous Metals 2013,23,2243. [王 敏,栾海燕,余 萍,车寅生,牛 超,董 多. 中国有色金属学报,2013,23,2243.] doi: 10.1016/S1003-6326(13)62724-7
(56)Rao,Z.; Gu,Y.; Huang,C. Y.; He,Y.; Huang,Y. P.; Zhang,A. Q. Environ. Chem. 2013,32,564. [饶 志,顾 彦,黄春迎,何 燕,黄应平,张爱清. 环境化学,2013,32,564.].
(57)Liu,Y.; Dai,C. H.; Ma,J. F.; Song,Z. W.; Sun,Y.; Fang,J. R.;Zgao,J. G.; Sun,X.; Gao,C.; Liu,Z. S. Bull. Chin. Ceram. Soc. 2008,28,1220. [刘 晔,戴长虹,马峻峰,宋祖伟,孙 勇,房晶瑞,赵金刚,孙 霞,高 敞,刘振森. 硅酸盐通报,2008,28,1220.]
(58)Liang,X.; Zhu,S.; Zhong,Y.; Zhu,J.; Yuan,P.; He,H.; Zhang,J. Appl. Catal. B 2010,97,151. doi: 10.1016/j.apcatb.2010.03.035
(59)Kaneti,Y. V.; Zhang,Z.; Yue. J.; Jiang,X.; Yu,A. J. Nanopart. Res. 2013,15,1948. doi: 10.1007/s11051-013-1948-z
(60)Kim,K.; Kim,I. H.; Yoon,K. Y.; Lee,J.; Jang,J. H. J. Mater. Chem. A 2015,3,7706. doi: 10.1039/C5TA00027K
欢迎订阅《大学化学》
经北京市新闻出版广电局批复(京新广函[2015]200号),自2016年1月起:《大学化学》(CN11-1815/O6)由双月刊变更为月刊。
《大学化学》是由教育部主管、北京大学和中国化学会共同主办的化学教育类刊物。主要介绍化学科学的新进展,开展与教学有关的重大课题的研讨,交流教学改革经验,报道化学及其相关学科的新知识、新动向,促进教师知识更新,扩大学生知识面,为提高教学水平服务。主要栏目有:今日化学、教学研究与改革、知识介绍、化学实验、师生笔谈、自学之友、大学化学先修课程、竞赛园地、国外化学教育、化学史、书评以及专题讨论等。
2016年每本定价12.00元,全年出版12期,共144.00元。
全国各地邮局均可订阅,邮发代号:82-314。为方便读者订阅,本刊编辑部全年办理邮购业务。
邮购地址:北京大学化学学院 《大学化学》编辑部
邮编:100871
电话:010-62751721
Email:dxhx@pku.edu.cn
网址:http://www.dxhx.pku.edu.cn
《大学化学》编辑部
Enhancement of the Photoelectrochemical Activity of α-Fe2O3Materials by Surface Modification with Vanadium
YAO Li-Zhen KONG De-Sheng*DU Jiu-Yao WANG Ze ZHANG Jing-Wei WANG Na LI Wen-Juan FENG Yuan-Yuan
(Department of Chemistry and Chemical Engineering,Qufu Normal University,Qufu 273165,Shandong Province,P. R. China)
Surface modification of semiconductor materials is an effective way to improve their photocatalysis and photo-conversion activities. Bare and V-modified α-Fe2O3photoelectrode materials were prepared using hydrothermal, chemical bath deposition and heat treatment approaches. Their physicochemical and photoelectrochemical (PEC) properties were then investigated with X-ray diffractometry(XRD), UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS), voltammetry, and electrochemical AC impedance spectroscopy (EIS) techniques. The existence of FeVO4was indicated by its characteristic X-ray diffractometry patterns, while no significant red shifts in the photoabsorption edge were detected in UV-Vis diffuse reflectance spectroscopy spectra. With V-modified and bare Fe2O3serving as a photoanode, photoelectrochemical measurements were carried out for water splitting in 1 mol·L-1NaOH (pH 13.6). The enhancement of α-Fe2O3photoelectrochemical activities through V-modification was indicated by significantly increased photocurrents and decreased photocharge-recombination probability. By measuring electrochemical AC impedance spectroscopy spectra, pseudo-first-order rate constants for the charge transfer at the illuminated electrode/solution interface were estimated. The rate constant for V-modification of the Fe2O3electrode was higher than that of the bare Fe2O3electrode. Improved interfacial charge transfer kinetics through V-modification is responsible for the enhanced photoelectrochemical activities of α-Fe2O3. The interfacial photocharge transfer and recombination processes and their properties are discussed with a semiconductor energy band model constructed for the electrode system.
Water splitting; Interfacial charge transfer; Surface recombination; Photocurrent;AC impedance spectroscopy
July 10,2015; Revised: September 4,2015; Published on Web: September 7,2015.
. Email: kongdscn@eyou.com; Tel: +86-537-4453069.
O649
10.3866/PKU.WHXB201509074
The project was supported by the Natural Science Foundation of Shandong Province,China (ZR2010EM026) and National Training Programs of Innovation and Entrepreneurship for Undergraduates,China (201410446044).
山东省自然科学基金(ZR2010EM026)和国家级大学生创新创业训练计划项目(201410446044)资助
猜你喜欢
物理通报(2024年4期)2024-04-09 12:41:28
中学生数理化·中考版(2021年10期)2021-11-22 07:26:40
人工晶体学报(2021年3期)2021-04-17 05:31:10
电子与封装(2021年3期)2021-03-29 06:31:28
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:07:02
中国资源综合利用(2016年4期)2016-01-22 08:27:22
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
新高考·高一物理(2015年6期)2015-09-28 20:10:57
电源技术(2015年2期)2015-08-22 11:28:02
山西大同大学学报(自然科学版)(2014年4期)2014-11-02 06:48:29