
凹形树突状PtCu纳米催化剂的合成及对甲醇的电催化
2015-09-15 01:40梅素娟吴俊杰陆双龙曹雪琴顾宏伟唐明华
无机化学学报 2015年12期
梅素娟 吴俊杰 陆双龙 曹雪琴 顾宏伟*, 唐明华
(1苏州大学材料与化学化工学部,苏州 215123)(2苏州大学分析测试中心,苏州 215123)
Pt纳米材料是电催化氧化有机小分子燃料电池最有效的电催化剂,包括直接甲醇燃料电池(DMFCs)。Pt纳米催化剂的电催化活性很大部分决定于它们的表面活性、粒子的形貌和尺寸以及它们的化学组成[1-2]。而且,纳米材料的形态或形状决定于它们的晶面的选择(或表面结构),电子结构,表面结合配体以及纳米粒子组装,这将直接影响到纳米材料的性能[3-8]。Sun课题组曾报道,具有高指数晶面的Pt纳米晶体对乙醇和甲酸的电催化氧化表现出很高的催化活性[9]。最近报道,Xia用两步法合成树突状Pd-Pt纳米材料[3]。这种特殊三维结构的Pt基树突状纳米材料在氧化还原反应中,比商业Pt/C催化剂具有更好的催化活性和稳定性。
根据Pt材料在电催化甲醇氧化反应中的良好性能,使它成为一种非常理想的直接甲醇燃料电池的电极材料[1,10-13]。这项技术商业化的主要限制之一是,在室温或适当的温度下,纯Pt材料易被反应过程中生成的中间有毒物种(CO)毒化[14-16],所以降低Pt纳米材料的毒化率是目前迫切需要解决的问题。提高催化剂抗毒性的一种方法是制备具有高指数晶面的纳米Pt材料[17],另一种方法是通过加入第二金属制成双金属的Pt合金催化剂[18-23]。此外,由于Pt的成本太高而且储存量有限,所以近年来研究的重点方向是通过研究具有高活性的Pt基纳米合金催化剂来降低Pt的使用量[24-26]。经研究发现,Pt通过与过渡金属的结合形成合金是增强Pt基催化剂活性的一种非常成功的方法[18,27]。近年来,一系列的纳米合金催化剂,包括 Pt-Ni[18]、Pt-Co[19]、Pt-Cu[20]、Pt-Sn[21]和Pt-Zn[22],作为低成本的直接甲醇燃料电池的阳极电极材料被报道。最近,Lou课题组通过简单的溶剂热法制备出在电催化甲醇氧化反应中具有较高催化活性的立方形的PtCu3纳米笼[23]。目前,大多数溶剂热法合成多孔状金属纳米材料都是两步法:首先形成固体纳米粒子,然后通过Kirkendall效应或者电迁移、生锈、刻蚀和腐蚀去合金方法得到空隙空间[23,28-34]。
由于Pt-Cu双金属纳米材料的高稳定性和强抗催化剂中毒性,以及自然界中丰富的铜资源,使得Pt-Cu双金属纳米材料成为甲醇燃料电池的电极材料最佳选择之一。不同形貌的Pt-Cu双金属纳米材料最近被大量的报道,如纳米立方体[15,35]、纳米球[36]、纳米笼[23]等。凹形树突状材料具有较大的比表面积和三维活性面等优势,也已大量研究[38]。因为高指数晶面上存在的高密度原子阶、边缘和节点,所以凹形Pt-Cu纳米材料在甲醇的电催化氧化反应中表现出非常好的催化活性[15]。研究发现通过电化学或者化学方法,Cu可以从合金的表层被移除,形成一种表面Pt密度较高的核壳结构。在甲醇的电催化氧化反应中,这种核壳结构的PtCu比纯Pt纳米粒子的催化活性高,抗催化剂中毒性强[23]。
在本文中,我们通过一锅法合成凹形树突状PtCu纳米催化剂:在邻苯二胺为表面活性剂,油胺为溶剂,乙酰丙酮铂(Pt(acac)2)、乙酰丙酮铜(Cu(acac)2)为前驱体的条件下,在160℃下水热釜中反应4 h。然后,我们将PtCu NCDs应用于甲醇的电催化氧化反应中,结果表明其具有很好的催化活性和稳定性。
1 实验部分
1.1 试剂与仪器
Pt(acac)2,Cu(acac)2和 Nafion 溶液(质量分数为5%)为Alfa Aesar公司的产品。20%的商业Pt/C催化剂在上海河森电气有限公司购买,其他试剂均购买于国药集团化学试剂有限公司,三次蒸馏水是自制的。
透射电镜 (TEM)用TecnaiG220和Tecani G2 F20(美国FEI公司)测试,加速电压为200 kV。X射线衍射(XRD)图在 X′Pert-Pro MPD-射线衍射仪(荷兰帕纳科公司) 上检测,Cu Kα,λ=0.154 059 8 nm,工作电压40 kV,电流40 mA。电感耦合等离子体原子发射光谱(ICP-AES)测试:美国VARIAN公司的Vista MPX仪器。X射线光电子能谱(XPS):Escalab 250iXL,Al Kα。电化学测试用CHI600电化学分析仪器 (美国CHI仪器公司)和常规的三电极体系进行。
1.2 PtCu NCDs的制备
9.87 mg Pt(acac)2,13 mg Cu(acac)2,6 mg 邻苯二胺和10 mL油胺,在常温下搅拌30 min,使它们很好的溶解在油胺中。然后,将得到的均相混合溶液转移至20 mL的水热釜中,在160℃的条件下反应4 h。最后,用正己烷分散,乙醇沉淀离心洗涤2~3次。真空干燥备用。
先取5 mg上述制备的纳米催化剂,加入1 mL异丙醇超声使纳米催化剂均匀分散,再加入25μL Nafion溶液(5%,w/w)和4 mL三次水,超声混合均匀,即可得到电催化剂油墨溶液。
1.3 工作电极的准备
依次用 1.0、0.3 和 0.05 μm 的氧化铝粉末在1 200目的金相砂纸 (灰色)、尼龙抛光布 (白色)、Microcloth抛光绒布(褐色)上把玻碳电极(Φ=3 mm)研磨抛光,用大量自来水冲洗,再分别用无水乙醇和超纯水超声洗涤3次,每次超声清洗时间不宜过长(大约在1 min),最后用超纯水冲洗干净,用N2吹干。 将电极插入到 0.5 mol·L-1H2SO4溶液中,在-0.2~1.4 V电位之间,以50 mV·s-1的扫描速度进行循环伏安扫描25 min,将电极表面进一步清洗,如果没有任何氧化还原峰出现,即可表明玻碳电极表面处理干净。再将已清洗干净的电极用超纯水冲洗干净后,用N2氛围下吹干备用。接着,取5μL上述所制备的电催化剂油墨溶液,滴加到玻碳电极上,并置于空气中自然干燥4 h,即得到电化学测量所需要的工作电极,为电化学实验做好准备。
1.4 电化学测试
所有电化学测试中所用的电解质溶液在进行电化学测量前均需要进行除氧准备,即通入30 min的N2。
在三电极体系中用循环伏安法测量催化剂的电化学活性比表面积(ECSAs),饱和甘汞电极(SCE)作为参比电极,铂片为辅助电极,玻碳载催化剂电极为工作电极,电解液为 0.5 mol·L-1H2SO4水溶液。为了除去催化剂表面残余污染物,首先将载有催化剂的工作电极置于 N2饱和的 0.5 mol·L-1H2SO4水溶液中,扫描范围为-0.2~1.1 V下循环扫描数次直到得到稳定的循环伏安(CV)曲线图为止,扫描速率为 50 mV·s-1。 然后,在该体系下,扫描范围为-0.25~1.0 V时记录扫描CV曲线图,根据H在催化剂上的电化学吸附曲线来计算催化剂的电化学活性比表面积。
在三电极体系中用循环伏安法、线性扫描法和计时电流法研究催化剂对甲醇的电催化氧化性能。电解液为 0.5 mol·L-1H2SO4+0.5 mol·L-1CH3OH。 循环伏安扫描范围为0.0~1.0 V;线性扫描法主要是为了比较催化剂对甲醇电催化氧化的起始氧化电位,扫描范围为0.0~0.6 V;计时电流法是在恒电位极化到0.5 V时测量甲醇电催化氧化电流随时间变化的曲线,保持15 min记录。以上所有测试条件均在室温条件,扫描速率为50 mV·s-1的条件下进行的。
2 结果与讨论
2.1 PtCu NCDs的物理表征
我们采用XRD技术研究已经制备出的PtCu NCDs和用相同方法制备出的Pt纳米材料的晶体结构。如图1所示,纯Pt纳米材料的Pt(111)、Pt(200)、Pt(220)和 Pt(311)的晶面衍射峰被检测出来,表明纯Pt纳米材料是一种面心立方结构的晶体材料。根据PtCu NCDs的XRD图中的特征峰的位置判断,没有明显的Pt、Cu的特征峰出现,并且相对于Pt的特征峰位置正移,与Cu的特征峰相比又发生负移,更加接近于PtCu合金的特征峰位置,从而证明PtCu NCDs是一种合金材料。
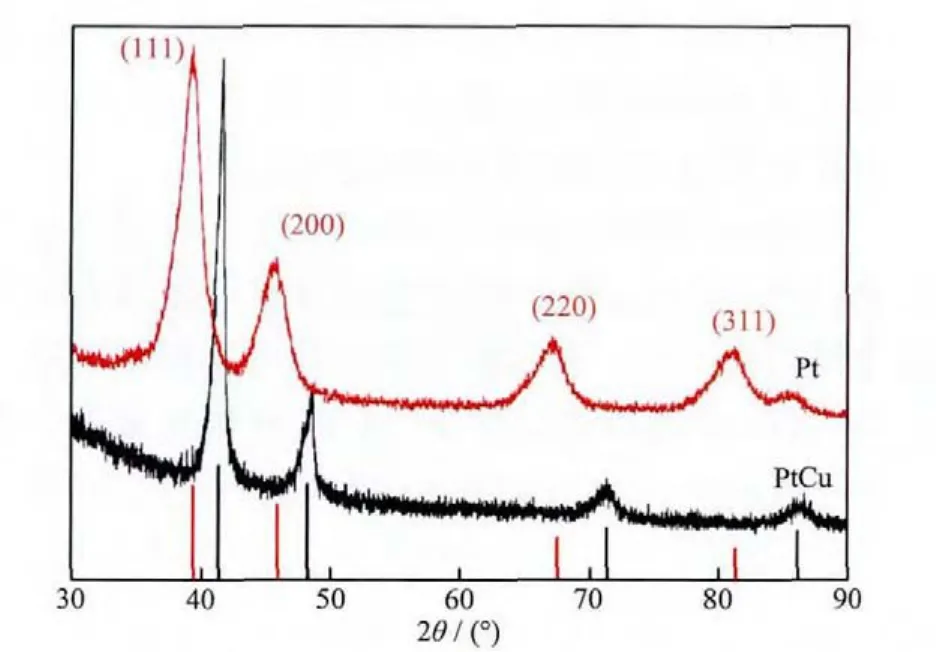
图1 PtCu NCDs与用同样方法合成的Pt纳米材料的XRD对比图Fig.1 XRD patterns of PtCu NCDs and Pt nanoparticles synthesized by the same method
图2 对应于PtCu NCDs的EDX谱图。EDX谱图的分析结果表明,PtCu NCDs是由Pt、Cu两种元素组成的,且Pt与Cu的原子数量比为1∶2.5。这与投料时加入的Pt、Cu前驱体的比例接近,表明加入的Pt和Cu几乎被完全还原出来。通过电感耦合等离子体测试制备的PtCu NCDs中Pt和Cu的实际含量,测试结果显示 Pt约为 30.2%,Cu 约为 69.8%,Pt与 Cu的原子比为1∶2.3,与EDX测试结果基本一致。
图S1为PtCu NCDs的X射线光电子能谱。图S1A中在74.1和 71.0 eV的峰分别对应于 Pt元素的4f5/2和4f7/2结合能[39]。图S1B中Cu2p3/2的结合能为 932.0 eV,相较于 Cu0(Cu2p3/2,932.7 eV)发生了负移,表明发生了原子间距的增大,从而证明PtCu NCDs是以合金形式存在的[40],与XRD的表征结果一致。此外,在933.6 eV处的峰和943.4 eV处出现的卫星信号峰说明有Cu2+的存在,因为Cu在潮湿空气中可被氧化[41]。

图2 PtCu NCDs的EDX谱图Fig.2 EDX Spectrum of PtCu NCDs
图3 A为PtCu NCDs的TEM图,图3B为PtCu NCDs的STEM图。从图3A和B中可以看出PtCu NCDs具有三维结构的凹形树突状材料,且有生成笼状结构的趋势。图3C为经过统计多个PtCu粒子后获得的粒径分布图。图中显示约有80%的PtCu粒子粒径分布在28~29 nm范围内,平均粒径为28.5 nm,表明通过水热釜法成功合成粒径分布较窄的PtCu NCDs。图3D为PtCu NCDs的高倍电镜图(HRTEM),显示出清楚地晶格条纹,晶面间距约为0.22和0.19 nm,这与面心立方结构的PtCu合金材料的(111)和(200)晶面间距一致。图3A中的插图为选择区域的电子衍射图,表明PtCu NCDs具有很高的结晶度。此外,图中可以清楚地看出PtCu NCDs的(111)、(200)、(220)和(311)晶面,与XRD的测量结果完全一致,更加证明了PtCu NCDs的面心立方结构。
邻苯二胺在此反应中起着至关重要的作用。当反应体系中不加入邻苯二胺时,得到图S2A中形貌不规则、尺寸不均一的PtCu材料;当邻苯二胺的加入量为6 mg时,如图S2B所示,得到形貌规则、尺寸均一的凹形树突状材料。因此,我们推测邻苯二胺在油胺中作为共还原剂,可能会影响Pt和Cu的还原速率。尽管CuⅡ/Cu(0.34 V)的标准还原电势小于 PtⅡ/Pt(1.18 V)的标准还原电势,Cu2+先被还原成Cu纳米晶体,然后Pt2+再与Cu纳米晶体进行电流置换反应得到凹形树突状的PtCu纳米材料[23,42]。然而,随着邻苯二胺的加入量增加到12 mg,纳米粒子的尺寸变得不均一,并且容易聚集(图S2C)。所以,我们推测邻苯二胺在体系中是作为结构导向剂和稳定剂存在的,适当的投入量能促进凹形树突状PtCu纳米材料的形成[43]。

图3 PtCu NCDs的TEM图 (A)、STEM图 (B)、粒径分布图 (C)和HRTEM图 (D)Fig.3 TEM(A),STEM(B),Particle size distribution(C)and High-resolution(HR)TEM(D)images of PtCu NCDs
2.2 PtCu NCDs对甲醇氧化的电化学性能
这种Cu元素含量较多的PtCu纳米材料作为电催化剂引起了广泛的兴趣。研究表明,Cu的选择性电化学溶解是活性电催化剂形成的关键步骤[23,36]。如图4A所示,在第二圈的循环伏安曲线中没有明显的 H吸附和 H脱附峰 (-0.25~0.1 V vs SCE),这可能是由于PtCu NCDs表面含有较多的Cu导致的。在第50圈的循环伏安曲线时,Cu溶解的电流密度减小,与此同时H的吸附和H的脱附峰逐渐出现。在50圈之后,Cu的溶解信号峰完全消失,得到一个稳定的与Pt类似的CV曲线。这种去合金过程可能导致催化剂的表面重排,得到表面含Pt较多的结构,达到优化催化剂某些性能的目的,如增大表面积等。图4B为PtCuNCDs和商业Pt/C催化剂在0.5 mol·L-1H2SO4溶液中的循环伏安曲线图。从图4B中可看出PtCu NCDs和商业Pt/C催化剂的氧化还原电势分别为0.54和0.49 V。PtCu NCDs的OHad吸附脱附峰比商业Pt/C催化剂的OHad吸附脱附峰正移了50 mV,表明OHad在PtCu NCDs表面的吸附能比商业Pt/C小,从而加速和氧气的氧化反应的进程[37]。电化学活性比表面积(ECSA)是衡量催化剂性能的重要参数之一,可以通过去除双电层后的H吸附或H脱附峰的积分面积计算。氧化一个单层H所需要的电量为0.21 mC·cm-2。根据图4B中的H吸附面积计算可得,PtCu NCDs的 ECSAs为 49.5 m2·g-1,比商业 Pt/C 的 ECSAs(48.7 m2·g-1)高。
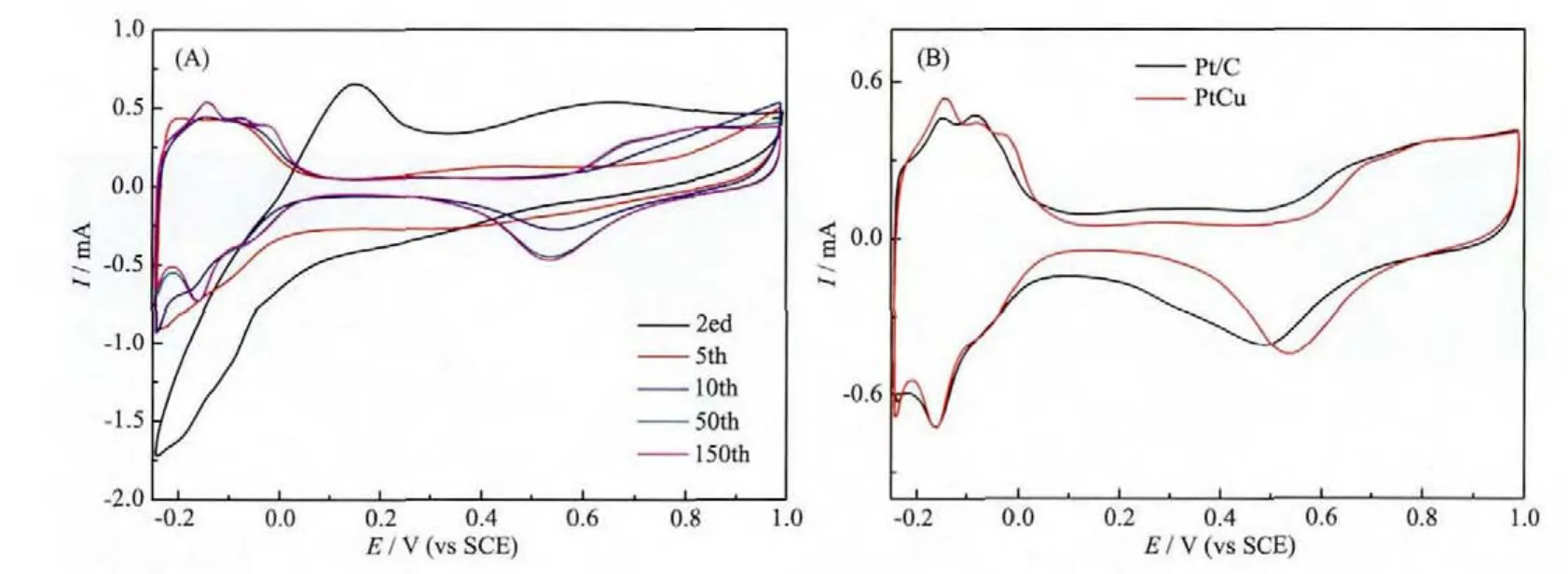
图4 (A)PtCu NCDs在扫描速率为50 mV·s-1时的循环伏安曲线图;(B)PtCu NCDs和商业Pt/C催化剂在N2-0.5 mol·L-1 H2SO4溶液中的循环伏安曲线图,扫描速率为50 mVs-1Fig.4 (A)Cyclic voltammetric(CV)profiles of PtCu NCDs at 50 mV·s-1;(B)CV profiles of PtCu NCDs and commercial Pt/Ccatalysts in a N2-0.5 mol·L-1 H2SO4 solution(50 mV·s-1)
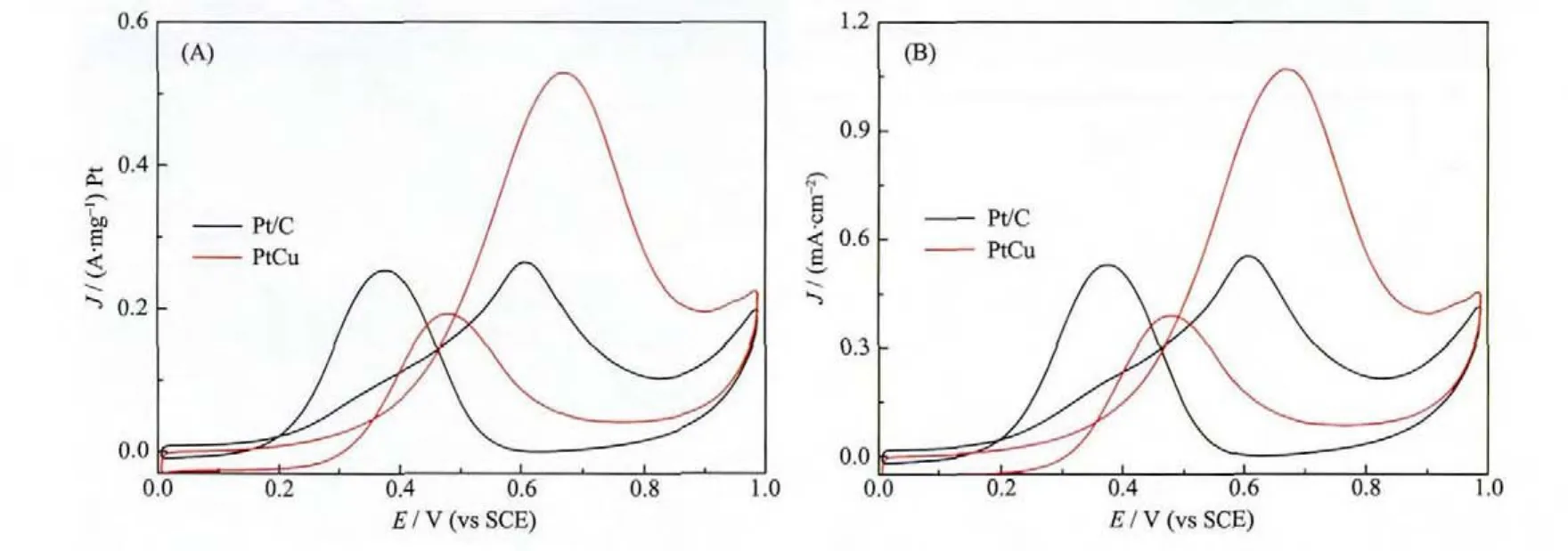
图 5 (A)PtCu NCDs和商业 Pt/C 电催化剂在 N2-0.5 mol·L-1 H2SO4+0.5 mol·L-1 CH3OH 的质量活性;(B)PtCu NCDs和商业 Pt/C 电催化剂在 N2-0.5 mol·L-1 H2SO4+0.5 mol·L-1 CH3OH 的比活性,扫描速率为 50 mV·s-1Fig.5 (A)Mass activites of methanol oxidation recorded in N2-0.5 mol·L-1 H2SO4+0.5 mol·L-1 CH3OH solution at a scan rate of 50 mV·s-1;(B)Specific activities of methanol oxidation recorded in N2-0.5 mol·L-1 H2SO4+0.5 mol·L-1 CH3OH solution at a scan rate of 50 mV·s-1
PtCu NCDs对甲醇的电催化氧化循环伏安(CV)测试图如图5所示。电解质为N2饱和的0.5 mol·L-1H2SO4+0.5 mol·L-1CH3OH水溶液体系,扫描速率为50 mV·s-1,室温下进行。我们用商业Pt/C作为对比催化剂。图5A和B分别对应于甲醇氧化的质量活性和比活性。从图5A中CV曲线可以看出,PtCu NCDs对于甲醇氧化的质量活性为0.53 A·mg-1Pt是商业Pt/C催化剂(0.26 A·mg-1Pt)的 2.04倍。从比活性的CV曲线图(图5B)对比发现PtCu NCDs(1.07 mA·cm-2)是商业 Pt/C催化剂(0.55 mA·cm-2)的1.95倍。而且,PtCu NCDs(2.76)比商业Pt/C催化剂(1.02)表现出更高的If/Ib比值,其中If是正扫方向的电流密度,Ib是负扫方向的电流密度。由此说明,相较于商业Pt/C催化剂而言,在正向扫描时PtCu NCDs能够更有效的实现对甲醇的电催化氧化反应,而且在负向扫描时产生较少的中间有毒物种(如CO等),从而降低了催化剂的毒化效应。由此说明PtCu NCDs具有很好的甲醇电催化活性和抗中毒性。

图6 PtCu NCDs和商业Pt/C电极在N2-0.5 mol·L-1 H2SO4+0.5 mol·L-1 CH3OH溶液中的计时电流曲线,应用电位为 0.6 VFig.6 Chronoamperometric curves(at 0.6 V)of methanol oxidation on PtCu NCDs and commercial Pt/C catalysts in N2-0.5 mol·L-1 H2SO4+0.5 mol·L-1 CH3OH solution
计时电流法(i-t)是检测电化学催化剂稳定性和抗毒性的重要手段。电解质为N2饱和的0.5 mol·L-1H2SO4+0.5 mol·L-1CH3OH水溶液体系,室温下进行实验。图6是将PtCu NCDs和商业Pt/C催化剂在电势为0.6 V时持续极化15 min记录得到的i-t曲线图。如果甲醇氧化的速度大于中间产物(如CO)的去除反应速度,中间物种将会在催化剂的表面不断积累,导致甲醇的氧化电流降低。根据图6中数据分析可得,PtCu NCDs和商业 Pt/C相比,PtCu NCDs对甲醇的电催化氧化的衰减速度更加缓慢。在连续氧化900 s后,PtCu NCDs对甲醇电催化氧化的峰电流密度为0.089 A·mg-1Pt,商业Pt/C催化剂对甲醇电催化氧化的峰电流密度为0.054 A·mg-1Pt。在整个甲醇氧化过程中,甲醇在PtCu NCDs上的峰电流密度始终维持在较高的位置上,这表明PtCu NCDs具有良好的稳定性和抗毒性能力。综上所述,与商业Pt/C催化剂相比,PtCu NCDs在甲醇电催化氧化反应中具有更高的活性,其原因可能为以下几点:(1)与商业的Pt/C催化剂的TEM(图S3)相比,PtCu NCDs具有较大表面积的凹形树突状材料更容易接近目标分子(如甲醇);(2)在各维度中相互连接的纳米结构提供了良好的电子导电性,从而导致电极表面的反应动力学加快;(3)PtCu合金的形成为Pt原子在纳米晶体表面提供合适的结构排列,如为甲醇氧化提供活性更高的晶面或合适的Pt-Pt原子间距[44-45]。因此PtCu NCDs的优异的电化学性能可能是因为其特殊凹形树突状的形貌和PtCu合金的形成。
3 结 论
本文采用水热釜法在有机溶液体系中一步合成PtCu NCDs。与商业Pt/C相比,PtCu NCDs在甲醇的电催化氧化反应中表现出很好的催化性能。PtCu NCDs在甲醇氧化反应中的优异性能特别是很强的抗CO中毒能力,可能归功于其独特的凹形结构和Pt、Cu组分的协同作用。这种特殊结构的PtCu NCDs是一种非常理想的MOR阳极材料。根据本文的实验结果推断出可以通过调控催化剂的结构和组成部分来优化催化剂的性能。
Supporting information is availableat http://www.wjhxxb.cn
[1]Chen J,Lim B,Lee E P,et al.Nano Today,2009,4(1):81-95[2]Peng Z,Yang H.Nano Today,2009,4(2):143-164
[3]Lim B,Jiang M,Camargo P H C,et al.Science,2009,324(5932):1302-1305
[4]Larsson E M,Alegret J,Kll M,et al.Nano Lett.,2007,7(5):1256-1263
[5]Alivisatos P.Nat.Biotechnol.,2004,22(1):47-52
[6]Huang X,El-Sayed I H,Qian W,et al.J.Am.Chem.Soc.,2006,128(6):2115-2120
[7]Mulvihill M J,Ling X Y,Henzie J,et al.J.Am.Chem.Soc.,2009,132(1):268-274
[8]Zhou K,Li Y.Angew.Chem.,Int.Ed.,2012,51(3):602-613
[9]Tian N,Zhou Z Y,Sun SG,et al.Science,2007,316(5825):732-735
[10]Guo S,Zhang S,Sun X,et al.J.Am.Chem.Soc.,2011,133(39):15354-15357
[11]Guo S,Dong S,Wang E.ACSNano,2009,4(1):547-555
[12]Wu H,Li H,Zhai Y,et al.Adv.Mater.,2012,24(12):1594-1597
[13]Chen A.Chem.Rev.,2010,110(6):3767-3804
[14]Yoo SJ,Jeon TY,Kim K S,et al.Phys.Chem.Chem.Phys.,2010,12(46):15240-15246
[15]Yin A X,Min X Q,Zhu W,et al.Chem.Eur.J.,2012,18(3):777-782
[16]Kugai J,Moriya T,Seino S,et al.Int.J.Hydrogen Energy,2012,37(6):4787-4797
[17]Huang X,Zhao Z,Fan J,et al.J.Am.Chem.Soc.,2011,133(13):4718-4721
[18]Stamenkovic V R,Fowler B,Mun B S,et al.Science,2007,315(5811):493-497
[19]Stamenkovic V R,Mun B S,Arenz M,et al.Nat.Mater.,2007,6(3):241-247
[20]Strasser P,Koh S,Anniyev T,et al.Nat.Chem.,2010,2(6):454-460
[21]Liu Y,Li D,Stamenkovic V R,et al.ACS Catal.,2011,1(12):1719-1723
[22]Kang Y,Pyo J B,Ye X,et al.ACSNano,2012,6(6):5642-5647
[23]Xia B Y,Wu H B,Wang X,et al.J.Am.Chem.Soc.,2012,134(34):13934-13937
[24]Gasteiger H A,Markovic N M.Science,2009,324(5923):48-49
[25]Gasteiger H A,Kocha SS,Sompalli B,et al.Appl.Catal.,B,2005,56(1):9-35
[26]Stephens I E L.Angew.Chem.,Int.Ed.,2011,50(7):1476-1477
[27]Greeley J,Stephens I E L.Nat.Chem.,2009,1(7):552-556
[28]Zeng J,Zhang Q,Chen J,et al.Nano Lett.,2009,10(1):30-35
[29]Yamauchi Y,Sugiyama A,Morimoto R,et al.Angew.Chem.,Int.Ed.,2008,47(29):5371-5373
[30]Prevo B G,Esakoff SA,Mikhailovsky A,et al.Small,2008,4(8):1183-1195
[31]Yin Y,Erdonmez C,Aloni S,et al.J.Am.Chem.Soc.,2006,128(39):12671-12673
[32]Schwartzberg A M,Olson T Y,Talley C E,et al.J.Phys.Chem.B,2006,110(40):19935-19944
[33]Wu Y,Wang D,Niu Z,et al.Angew.Chem.,Int.Ed.,2012,51(50):12524-12528
[34]Yavuz M S,Cheng Y,Chen J,et al.Nat.Mater.,2009,8(12):935-939
[35]Xu D,Liu Z,Yang H,et al.Angew.Chem.,Int.Ed.,2009,48(23):4217-4221
[36]Koh S,Strasser P.J.Am.Chem.Soc.,2007,129(42):12624-12625
[37]Kibsgaard J,Gorlin Y,Chen Z,et al.J.Am.Chem.Soc.,2012,134(18):7758-7765
[38]Liu H,Nosheen F,Wang X.Chem.Soc.Rev.,2015,44(10):3056-3078
[39]Tseng Y C,Chen H S,Liu C W,et al.J.Mater.Chem.A,2014,2(12):4270-4275
[40]Shiraishi Y,Sakamoto H,Sugano Y,et al.ACSNano,2013,7(10):9287-9297
[41]Xu C,Liu Y,Wang J,et al.J.Power Sources,2012,199:124-131
[42]Zhang Z,Yang Y,Wosheen F,et al.Small,2013,9(18):3063-3069
[43]Jin R C,Cao Y W,Mirkin C,et al.Science,2001,294(5548):1901-1903
[44]Mohanty A,Garg N,Jin R C.Angew.Chem.Int.Ed.,2010,49(29):4962-4966
[45]Lim B,Lu X,Jiang M,et al.Nano Lett.,2008,8(11):4043-4047
猜你喜欢
中学生数理化·中考版(2022年12期)2022-02-16
今日农业(2019年15期)2019-09-03
中国有色金属学报(2018年2期)2018-03-26
三联生活周刊(2017年43期)2017-10-20
环境保护与循环经济(2017年1期)2017-09-26
中学生理科应试(2017年2期)2017-04-01
中南大学学报(自然科学版)(2016年2期)2017-01-19
肇庆学院学报(2016年5期)2016-03-11
中国资源综合利用(2016年7期)2016-02-03
船舶标准化工程师(2015年3期)2015-11-18
