
痛风发病机制研究进展
2015-07-31 23:45董鹏,宋慧
基础医学与临床 2015年12期
董 鹏,宋 慧
(北京积水潭医院 北京大学第四临床医学院 风湿免疫科, 北京 100035)
短篇综述
痛风发病机制研究进展
董 鹏,宋 慧*
(北京积水潭医院 北京大学第四临床医学院 风湿免疫科, 北京 100035)
痛风是一种由于单钠尿酸盐(MSU)晶体与组织微环境相互作用导致的反复发作的急性炎性反应。MSU晶体启动机体的炎性级联反应涉及多种信号通路,其中包括MSU晶体直接与细胞表面Toll样受体的结合,MSU晶体与细胞膜表面脂类分子的结合以及MSU晶体以内吞的方式进入细胞。NLRP3炎性小体的活化处于炎性反应的中心环节。
痛风;单钠尿酸盐;炎性反应;炎性小体
痛风是由于嘌呤类物质代谢紊乱,以致尿酸产生过多和(或)尿酸排泄减少,血尿酸浓度持续增高,最终导致尿酸盐结晶沉积软组织所致的一组代谢性疾病。痛风可分为原发性痛风和继发性痛风。原发性痛风由遗传因素和环境因素共同致病,具有一定的家族易感性,绝大多数病因未明。继发性痛风发生在其他疾病(如肾脏病、血液病等)过程中,或由服用某些药物、肿瘤放射治疗、化学治疗等多种原因引起。痛风早在公元前2640年就被古埃及人首次发现,并且在公元前5世纪被希波克拉底描述为“不可行走的疾病”[1]。随着社会经济的发展,痛风的发病率明显升高。然而截至目前,痛风的发病机制尚不完全清楚,本文主要就原发性痛风发病机制及相关研究进展进行概述,并对当下痛风机制研究中存在的一些问题进行探讨。
1 痛风临床表现
根据高尿酸血症和痛风发作的时间关系, 痛风一般可分为无症状高尿酸血症期、痛风间歇发作期和慢性痛风石病变期3个阶段[2]。痛风急性发作前
可无先兆,手术、高嘌呤饮食的摄入以及降尿酸治疗是诱发痛风发作的常见原因[3]。急性发作期缓解后,多数患者在初次发作后1~2年内复发。发作次数逐渐增多,症状持续时间延长,无症状间歇期缩短,受累关节逐渐增多。大量MSU晶体沉积于皮下、关节滑膜、软骨、骨质及关节周围软组织形成痛风石。近期研究发现在痛风急性发作之前,便可以在部分患者体内检测到MSU晶体的沉积和慢性炎症的存在,并且在急性发作期之后仍然存在。为了与单纯高尿酸血症相区别,对痛风的发展阶段进行了重新界定,包括:高尿酸血症无MSU晶体沉积期、有MSU晶体沉积但无相关临床症状期、伴有MSU晶体沉积的急性发作期、慢性痛风石病变期[4]。除了关节受损之外,长期的高尿酸血症或痛风患者常伴有心血管疾病和肾脏的损伤,因此,痛风从一开始就应该按照慢性疾病对待[3]。
2 痛风发病机制
痛风发病机制涉及尿酸代谢、尿酸盐晶体形成以及尿酸盐晶体与机体局部微环境相互作用最终诱发炎性反应等多个阶段。
2.1 尿酸代谢途径
尿酸是体内嘌呤代谢的产物,也是高尿酸血症和痛风发生的物质基础。人类由于进化过程中基因突变导致尿酸酶缺失,使得尿酸不能进一步水解成可溶性的尿囊素,从而导致血中尿酸水平升高。在人体内,尿酸本身不仅具有强大的抗氧化作用,而且担负着血清中50%以上自由基的清除任务,保护维生素C避免氧化,同时尿酸对维持人类血压直接相关。如图1所示,外源性和内源性嘌呤经肝脏代谢形成尿酸,尿酸在血清中以尿酸盐的形式存在,在体内循环形成“尿酸池”,主要经过肾脏以及肠道排泄, 其中肾脏排泄约2/3,肠道排泄约占1/3,血清尿酸平衡取决于尿酸生成与排泄的平衡[5]。
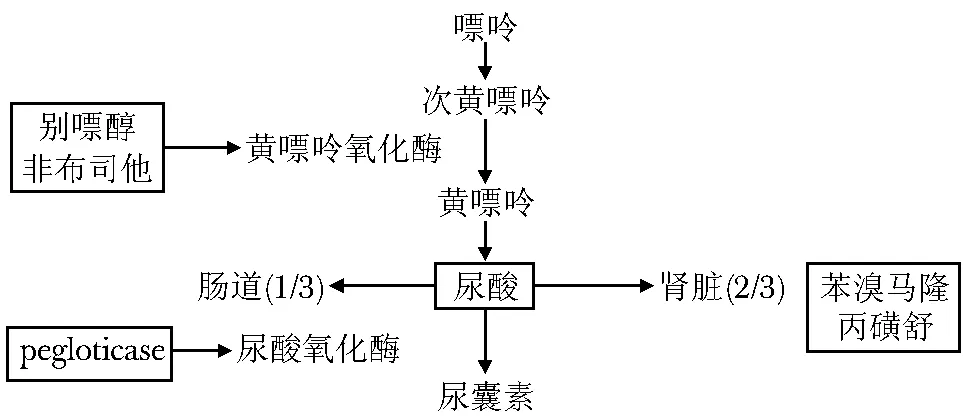
图1 嘌呤代谢途径及降尿酸药物相关靶点Fig 1 Metabolic pathway of purine and the targets of urate-lowering drugs
2.2 尿酸盐晶体的形成机制
尿酸盐晶体的形成是痛风发病机制的核心环节,然而在关节炎症部位血尿酸如何形成尿酸盐晶体的机制尚不完全清楚。当血清尿酸水平超过其物理溶解度(约7 mg/dL)时便以MSU晶体的形式析出。除此之外,溶质的pH值、温度、离子强度以及血浆中与尿酸结合的微粒都可以影响尿酸盐晶体的析出。与此同时,血清中的白蛋白可能作为尿酸盐晶体形成的核心单元。体液循环中的抗体蛋白,包括IgG及IgM也可以识别尿酸盐晶体的表面,进而起到稳定尿酸盐晶体并促进其进一步结晶的作用。早期研究认为,痛风的急性发作是由于尿酸盐晶体在关节部位瞬间形成所诱发的炎性反应。而最近的研究表明,在痛风急性发作之前,便可以在部分患者体内检测到MSU晶体的沉积和慢性炎症的存在[6,7],并且在急性发作期结束之后仍然存在[8]。这一发现与尿酸盐结晶过程较慢以及高尿酸血症患者往往需要较长时间才会出现痛风发作的现象一致[2]。
2.3 痛风发病的免疫分子机制
痛风急性发作是尿酸盐晶体与局部微环境相互作用诱发炎性反应的结果,其中涉及机体的固有免疫应答和适应性免疫应答机制,而前者发挥主导作用。固有免疫是人体免疫系统的第一道防线,具有反应启动快、无免疫记忆等特点。同时,部分固有免疫细胞可作为抗原提呈细胞进一步激活机体的适应性免疫应答。机体可以通过识别病原体相关分子模式(PAMPs)和危险信号相关分子模式(DAMPs)两种方式识别尿酸盐晶体,活化固有免疫细胞进而启动机体的固有免疫系统,其中涉及的固有免疫细胞包括单核/巨噬细胞、中性粒细胞、肥大细胞以及树突状细胞。然而尿酸盐晶体如何与固有免疫细胞直接相互作用并启动机体固有免疫系统,其机制尚不完全明确,可能涉及多条与炎症相关的信号通路[9- 10],以下将逐一进行介绍(图2)。
2.3.1 尿酸盐晶体直接与细胞表面的受体结合:巨噬细胞以及分化为巨噬细胞的单核细胞在痛风急性发作早期发挥主要作用[11]。Toll样受体(Toll-like receptors, TLR)是参与固有免疫的一类重要蛋白质分子,也是连接固有免疫和适应性免疫应答的桥梁。TLR是单个的跨膜非催化性蛋白质,作为一类经典的模式识别分子,TLR可以识别多种来源于微生物的具有保守结构的分子,如革兰阴性菌脂多糖(LPS)、CpG-DNA和ds-RNA等。如图2所示,尿酸盐晶体可以直接与单核巨噬细胞表面的TLR2和TLR4结合,进而通过MYD88接头分子活化单核/巨噬细胞内的多种转录因子如NF-κB,进而表达包括TNF-α、IL- 1β、IL- 6及IL- 8在内的多种细胞因子[10,12]。这些细胞因子可以进一步刺激内皮细胞表达包括E选择素在内的多种黏附分子上调,进而招募更多的中性粒细胞聚集于尿酸盐结晶沉积部位。尿酸盐晶体可以直接刺激招募而至的中性粒细胞内的酪氨酸激酶发生磷酸化,进而产生大量超氧负离子,这些超氧负离子可以介导炎性小体的组装和中性粒细胞的活化。活化的中性粒细胞进而又可以通过发生自噬以及分泌大量IL- 1β等炎性因子加剧尿酸盐沉积部位的炎性反应,进而将机体的炎性反应进一步放大[13]。

图2 单钠尿酸盐晶体通过多条信号通路活化固有免疫细胞
2.3.2 尿酸盐晶体以吞噬的方式进入细胞:巨噬细胞可以直接吞噬尿酸盐晶体,同时产生大量的活性氧分子(ROS),而活性氧分子可以直接活化炎性小体[14- 15]。于此同时,被吞噬的尿酸盐晶体可以导致溶酶体的肿胀、破坏,溶酶体的肿胀、破坏自身可以作为一种细胞内的危险信号引起细胞内钾离子的内流,而钾离子的内流即可直接活化炎性小体[16- 17]。近期研究发现,微管蛋白在炎性小体的组装和活化过程中起核心作用[18]。炎性小体是存在于髓系细胞内的一种由caspase- 1、caspase- 5、NLRP和PYCARD等多种蛋白分子组成的蛋白复合体,是固有免疫的重要组成部分,参与机体多种炎性反应的活化过程。近来发现这也是痛风急性发作的核心环节[19],其中研究最为清楚的尚属NLRP3炎性小体。NLRP3炎性小体由NLRP3、接头蛋白ASC以及caspase- 1组成。活化的caspase- 1可以切割IL- 1β和IL- 18的前体,产生相应的成熟细胞因子[20]。与正常人相比,痛风患者体内的IL- 1水平明显升高,而且急性痛风患者的体内的IL- 1水平远远高于慢性痛风患者[21]。新近出现的IL- 1抑制剂,如:anakinra、 canakinumab和rilonacept,在治疗痛风急性发作的方面的有效性,进一步证明了IL- 1在痛风急性发作过程中的核心作用[22- 24]。秋水仙碱也可以通过抑制炎性小体活化的多个节点,起到治疗痛风急性发作的作用[9]。
2.3.3 尿酸盐晶体与细胞膜的脂类分子直接接触:尿酸盐晶体可以不依赖细胞表面受体,直接与树突状细胞的细胞膜表面的脂类分子相结合(图2)。进一步研究发现尿酸盐晶体不能与细胞表面的磷脂酰胆碱和磷脂酰乙醇胺结合,而是与细胞膜的胆固醇成分直接结合;而且这种结合力度足够强大,可以通过干扰细胞表面脂类分子的迁移向细胞内部传递危险信号,进而引起酪氨酸激酶Syk的磷酸化,进一步引起PI3K激酶的活化,最终活化树突状细胞[25]。
除此之外,尿酸盐晶体还可以直接与补体系统的组成成分,如C1q、C1r、C1s、IgG和IgM结合,通过经典途径和旁路途径激活补体系统。补体系统活化可以产生大量的C5a和C5b等趋化因子,进而招募更过的淋巴细胞到达炎性反应部位。C6介导产生的膜攻击复合物可以进一步促进IL- 8的分泌以及炎症部位中性粒细胞的招募[9]。
2.3.4 痛风缓解的免疫调节机制:痛风发作症状一般在数天内便可迅速缓解,即使发病部位依旧可以检测到尿酸盐晶体的存在,而且已有痛风石形成的慢性痛风患者体内,痛风症状也并非持续发作。有关这一痛风免疫调节机制尚不完全清楚,可能涉及多种免疫调节细胞和免疫调节分子的参与。中性粒细胞胞外诱捕网(neutrophil extracellular traps,NETs)是由中性粒细胞释放到胞外的包含有核酸、组蛋白、髓过氧化物酶、中性粒细胞弹性蛋白酶和组织蛋白酶G等成分组成的网状结构,是新近发现的一种杀伤病原菌和防治病原菌扩散的新机制。研究发现,尿酸及尿酸盐晶体可以直接诱导中性粒细胞胞外诱捕网的形成,且部分依赖NF-κB信号通路的参与[26]。招募至痛风关节炎性反应部位的中性粒细胞被尿酸盐晶体刺激后释放大量的中性粒细胞胞外诱捕网,进而形成中性粒细胞胞外诱捕网交联体,这些交联体可以进一步通过丝氨酸蛋白酶作用降解炎症部位的炎性细胞因子和中性粒细胞趋化因子,进而中断炎性反应的恶性循环,起到负调节痛风关节炎炎性反应的作用[27]。但中性粒细胞属于痛风关节炎炎性反应中后期的效应细胞,有关痛风关节炎起始阶段的免疫调节细胞及相关调节分子研究尚未见报道。
3 痛风的药物治疗
痛风的药物治疗主要包括两方面内容,一方面控制血尿酸水平,另一方面控制痛风急性发作。如图1所示,目前市面主要存在3类降尿酸药物。第一,通过黄嘌呤氧化酶的抑制剂来减少尿酸的形成,代表药物如别嘌醇和非布司他。第二,通过促进尿酸排泄的药物增加肾脏对尿酸的排泄,代表药物如苯溴马隆和丙磺舒。第三,通过人工重组合成的尿酸氧化酶使尿酸代谢成为更加溶于水的尿囊素,代表药物,如pegloticase[28]。传统用以缓解痛风急性发作的药物包括秋水仙碱、非甾体药物以及糖皮质激素等。然而新近出现的IL- 1抑制剂,如anakinra、 canakinumab和rilonacept,在治疗痛风急性发作的效果也日益受到人们的关注。
4 痛风相关研究中存在的关键问题
随着中国经济的发展,人们生活方式和饮食结构的改变,人类寿命的延长,人口老龄化以及某些药物如利尿剂的广泛使用使得高尿酸血症和痛风发病率逐年升高。本文主要就当前痛风的发病机制及相关研究进行综述,虽然人们对这一古老疾病的了解较前明显增加,然而多个现实问题始终没有答案。例如:为何许多高尿酸血症患者终生没有痛风发作?而且即使在某些患者的身体局部检测到尿酸盐结晶的存在,这些患者始终没有痛风发作?到底是什么因素打破了局部微环境的免疫平衡,诱发了痛风的急性发作?当下的研究主要集中于固有免疫系统在痛风发病过程中的作用,而有关免疫调节机制如免疫调节细胞及相关细胞因子在痛风发病过程中的作用研究很少。针对这些问题的解决将会大大提高人们对痛风的预防和治疗能力。
[1] Fodor D, Nestorova R, Vlad V,etal. The place of musculoskeletal ultrasonography in gout diagnosis [J]. Med Ultrason, 2014, 16: 336- 344.
[2] Bardin T, Richette P. Definition of hyperuricemia and gouty conditions [J]. Curr Opin Rheumatol, 2014, 26: 186- 191.
[3] Perez-Ruiz F, Castillo E, Chinchilla SP,etal. Clinical manifestations and diagnosis of gout [J]. Rheum Dis Clin North Am, 2014, 40: 193- 206.
[4] Dalbeth N, Stamp L. Hyperuricaemia and gout: time for a new staging system? [J] Ann Rheum Dis, 2014, 73: 1598- 1600.
[5] de Oliveira EP, Burini RC. High plasma uric acid concentration: causes and consequences [J]. Diabetol Metab Syndr, 2012, 4: 12- 18.
[6] Pineda C, Amezcua-Guerra LM, Solano C,etal. Joint and tendon subclinical involvement suggestive of gouty arthritis in asymptomatic hyperuricemia: an ultrasound controlled study [J]. Arthritis Res Ther, 2011, 13: R4.
[7] De Miguel E, Puig JG, Castillo C,etal. Diagnosis of gout in patients with asymptomatic hyperuricaemia: a pilot ultrasound study [J]. Ann Rheum Dis, 2012, 71: 157- 158.
[8] Peiteado D, De Miguel E, Villalba A,etal. Value of a short four-joint ultrasound test for gout diagnosis: a pilot study [J]. Clin Exp Rheumatol, 2012, 30: 830- 837.
[9]Dalbeth N, Lauterio TJ, Wolfe HR. Mechanism of action of colchicine in the treatment of gout [J]. Clin Ther, 2014, 36: 1465- 1479.
[10] Busso N, So A. Microcrystals as DAMPs and their role in joint inflammation [J]. Rheumatology (Oxford), 2012, 51: 1154- 1160.
[11] Martin WJ, Shaw O, Liu X,etal. Monosodium urate monohydrate crystal recruited noninflammatory monocytes differentiate into M1-like proinflammatory macrophages in a peritoneal murine model of gout [J]. Arthritis Rheum, 2012, 63: 1322- 1332.
[12] Liu-Bryan R, Scott P, Sydlaske A,etal. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation [J]. Arthritis Rheum, 2005, 52: 2936- 2946.
[13] Mitroulis I, Kambas K, Chrysanthopoulou A,etal. Neutrophil extracellular trap formation is associated with IL- 1beta and autophagy-related signaling in gout [J]. PLoS One, 2011, 6: e29318.doi:10.1371/journal.pone.0029318.
[14] Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation [J]. J Clin Immunol, 2010, 30: 628- 631.
[15] Dostert C, Petrilli V, Van Bruggen R,etal. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica [J]. Science, 2008, 320: 674- 677.
[16] Hornung V, Bauernfeind F, Halle A,etal. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization [J]. Nat Immunol, 2008, 9: 847- 856.
[17] Petrilli V, Papin S, Dostert C,etal. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration [J]. Cell Death Differ, 2007, 14: 1583- 1589.
[18] Misawa T, Takahama M, Kozaki T,etal. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome[J]. Nat Immunol, 2013, 14: 454- 460.
[19] Mariathasan S, Newton K, Monack DM,etal. Differential activation of the inflammasome by caspase- 1 adaptors ASC and Ipaf [J]. Nature, 2004, 430: 213- 218.
[20] Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation [J]. Nat Rev Mol Cell Biol, 2003, 4: 95- 104.
[21] Qing YF, Zhang QB, Yang QB,etal. Altered expression of NLRP3 inflammasome in peripheral blood from gout patients might be associated with gouty arthritis [J]. Gout and Hyperuricemia, 2014, 1: 25- 32.
[22] Rees F, Hui M, Doherty M. Optimizing current treatment of gout [J]. Nat Rev Rheumatol, 2014, 10: 271- 283.
[23] Edwards NL, So A. Emerging therapies for gout [J]. Rheum Dis Clin North Am, 2014, 40: 375- 387.
[24] Mitha E, Schumacher HR, Fouche L,etal. Rilonacept for gout flare prevention during initiation of uric acid-lowering therapy: results from the PRESURGE- 2 international, phase 3, randomized, placebo-controlled trial[J]. Rheumatology (Oxford), 2013, 52: 1285- 1292.
[25] Ng G, Sharma K, Ward SM,etal. Receptor-independent, direct membrane binding leads to cell-surface lipid sorting and Syk kinase activation in dendritic cells[J]. Immunity, 2008, 29: 807- 818.
[26] Arai Y, Nishinaka Y, Arai T,etal. Uric acid induces NADPH oxidase independent neutrophil extracellular trap formation [J]. Biochem Biophys Res Commun, 2014, 443: 556- 561.
[27] Schauer C, Janko C, Munoz LE,etal. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines [J]. Nat Med, 2014, 20: 511- 517.
[28] Stamp LK, Merriman TR, Barclay ML,etal. Impaired response or insufficient dosage? Examining the potential causes of “inadequate response” to allopurinol in the treatment of gout [J]. Semin Arthritis Rheum, 2014, 44: 170- 174.
Research progress in mechanisms of gout
DONG Peng, SONG Hui*
(Dept. of Rheumatology and Immunology, Beijing Jishuitan Hospital, 4th Medical College of Peking University, Beijing 100035, China)
Gout is a disease characterized by recurrent attacks of acute inflammation which is triggered by interactions between endogenous monosodium urate (MSU) crystals and the local tissue environment. The mechanism by which MSU crystals activate pro-inflammatory cells involves several signal pathways. It is described that MSU crystals could bind to Toll-like receptors. MSU crystals may also interact with cholesterol on plasma membranes. Moreover, MSU crystals phagocytosis plays a critical role in the development of acute inflammation. Recent work has implicated the activation of NLRP3 inflammasome is the core step of acute inflammation.
gout; monosodium urate; inflammation; inflammasome
2015- 05- 11
2015- 10- 16
1001-6325(2015)12-1695-05
R971+.1
A
*通信作者(corresponding author):jst_fsmy@126.com
猜你喜欢
茶道(2022年3期)2022-04-27
中老年保健(2021年8期)2021-08-24
基层中医药(2021年5期)2021-07-31
昆明医科大学学报(2021年6期)2021-07-31
基层中医药(2021年1期)2021-07-22
天津医科大学学报(2021年3期)2021-07-21
华人时刊(2019年15期)2019-11-26
基层中医药(2018年8期)2018-11-10
基层中医药(2018年8期)2018-11-10
医学研究杂志(2015年9期)2015-07-01
