
应用改良连锁分析初筛家族性高胆固醇血症的候选基因
2015-07-25 05:01:10张筠婷王绿娅
中国全科医学 2015年18期
张筠婷,王绿娅
本研究创新点:
家族性高胆固醇血症 (FH)是一种异质性疾病,有多种致病基因,连锁分析作为致病基因的初筛方法之一,已被美国、丹麦、荷兰等多个国家广泛使用,而在我国并未广泛推广。其主要将基因组中与基因突变部位紧邻的多态性位点作为标记,利用凡带有该标记的成员可能携带致病基因的原理,对致病基因做出间接诊断。本研究在经典连锁分析基础上,结合新的微卫星位点和新型毛细管电泳仪,对FH进行基因初筛,针对2个FH家系,得出致病基因,为后期的测序工作提供方向。
家族性高胆固醇血症 (familial hypercholesterlolemia,FH)是一种较为常见的常染色体显性遗传性疾病,是单基因遗传性高胆固醇血症最常见的形式[1]。该病的杂合子发病率为1/500,纯合子较为罕见仅为1/1 000 000。这种遗传性的胆固醇代谢障碍通常导致过早发生动脉粥样硬化而早发冠心病[2]。其主要临床表现为血浆总胆固醇 (TC),特别是低密度脂蛋白胆固醇(LDL-C)水平显著升高,皮肤肌腱黄色瘤并伴有瓣膜反流等[3]。较其他类型的高脂血症而言,FH患者出现心血管疾病的临床表现更严重、更多样,其危害性也更大。FH研究成为一大热点,但我国对FH的研究报道较少。1974年 Goldstein等[4]提出,FH发病是由于低密度脂蛋白受体 (LDL-R)基因突变,引起细胞膜表面的LDL-R缺如或结构功能异常,导致体内低密度脂蛋白 (LDL)清除障碍并在组织内过度淤积。随着检测手段的快速发展,目前已经在世界各地发现了共1 741种突 变 (http://www.ucl.ac.uk/fh)。Jones 等[5]认 为,FH的病因不应仅考虑LDL-R基因突变,其他基因的突变也可导致典型的FH样表型。在已排除LDL-R基因突变的患者中,已经检测到多种致病基因,包括载脂蛋白B100(apo-B100)基因和新发现的枯草溶菌素9(PCSK9)基因等。
目前已有报道显示,LDL-R、apo-B100、PCSK9基因等多种基因突变均可导致FH[4-5],因此应采用一种快速、有效的基因筛查方法对FH患者进行初筛,尽早明确FH患者与各种基因的相关性。连锁分析作为遗传学初筛的主要方法,已经被美国、丹麦、荷兰等国家广泛使用[6],国外针对FH的主要致病基因,即LDL-R基因以及新发现的PCSK9基因,采用家系连锁分析的方法做了大量研究[7]。本研究也选用基因连锁分析的方法进行研究,并采用新的微卫星位点和新型毛细管电泳仪,对经典的连锁分析方法有所突破,最终为后期的研究奠定了基础。
1 对象与方法
1.1 研究对象 先证者1:男性,10岁,河南人,2010年6月以“高脂血症”入院。身上多部位黄色瘤,1岁时发现高脂血症,3岁时心功能Ⅱ级〔纽约心脏病协会 (NYHA)分级〕。6岁开始使用阿托伐他汀钙片(立普妥)进行降脂治疗。心功能逐渐恶化,身上肌腱部位黄色瘤逐年增大。有黄色瘤家族史、冠心病家族史。
先证者2:男性,8岁,北方人,2010年7月以“高脂血症”入院。身体多部位黄色瘤,如双手腕、双侧跟腱、臀部等,并出现角膜弓;血清TC水平为23.00 mmol/L,LDL-C 水平为 20.10 mmol/L。其表姨妈生前患有严重的高胆固醇血症,且皮肤多部位存在黄色瘤,已于15岁时患急性心肌梗死死亡。
根据陈在嘉等[8]主编的《临床冠心病学》提出的FH诊断标准:成人TC>7.80 mmol/L,16岁以下儿童TC>6.70 mmol/L或成人LDL-C>4.90 mmol/L以及患者或亲属有黄色瘤诊断为 FH。其中,TC>16.00 mmol/L并有黄色瘤者诊断为纯合子FH。2例先证者临床均符合FH的诊断。
1.2 研究方法
1.2.1 临床资料收集 所有家系成员进行血脂测定、心电图、超声心动图及颈动脉超声检查。记录家系成员的其他临床资料 (年龄、性别、发病年龄、病程、家族史及治疗情况)。患儿家属均签署知情同意书,收集家系成员空腹12 h外周血3 ml,乙二胺四乙酸二钠(EDTA-Na2)抗凝,血浆用于血脂测定,血细胞用于DNA提取。
1.2.2 临床检查 采用氧化酶法测定血浆中TC、三酰甘油 (TG)水平;磷钨酸镁沉淀及酶法测定高密度脂蛋白胆固醇 (HDL-C)水平;免疫沉淀法测定apo-B100水平;LDL-C按 Friedwald公式计算,LDL-C=TC-(HDL-C+TG/5)。对家系中所有成员进行生化检查。
1.2.3 致病基因突变检测
1.2.3.1 apo-B100基因Q3500R位点的检测 提取先证者基因组DNA,扩增apo-B100基因3500区域的片断309 bp。琼脂糖凝胶电泳鉴定,对鉴定后的PCR产物经正反双向核苷酸序列分析及与正常序列比对,排除由apo-B100基因3500附近位点突变导致的家族性apo-B100缺陷症 (FDB)。
1.2.3.2 连锁分析初筛各家系的易患基因 根据文献报道,微卫星位点 D1S417、D1S2797、D1S2890与PCSK9基因连锁;微卫星位点D19S221、D19S394与LDL-R基因连锁[9]。用微卫星短串联重复序列 (STR)扩增体系进行DNA定量及PCR,PCR后纯化,纯化后上机扫描,用GeneMarker读取峰值,在GeneMarker中导入家系文件,Genehunter软件和Lingkage软件处理并计算 LOD值。LOD值 >2.00,肯定连锁;LOD值<1.00,肯定不连锁;LOD 值1.00 ~2.00,可能连锁。
1.2.3.3 核苷酸序列分析 经琼脂糖凝胶电泳鉴定后符合目的片断长度的PCR产物,送交生工生物工程(上海)股份有限公司进行酶切、胶回收,进行核苷酸序列分析。将测序结果进行分析,找出突变位点;分析突变引起的氨基酸改变情况;检索FH突变数据库[10],如果在已经公布的1 741种突变中未见,则认为是新的突变位点。
1.3 统计学方法 采用SPSS 19.0统计学软件进行统计处理,计量资料以 (±s)表示,两组间比较采用t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 患儿心电图及超声检查结果 先症者1心电图示左心室肥厚;先症者2心电图大致正常 (见图1)。先证者1超声心动图示主动脉瓣反流;先证者2超声心动图示左房室瓣反流 (见图2)。先证者1颈动脉超声示颈动脉内中膜增厚,颈动脉内斑块形成;先证者2颈动脉超声示颈动脉内中膜增厚 (见图3)。

图1 先症者心电图结果Figure 1 ECG of probands

图2 先症者超声心动图结果Figure 2 UCG of probands
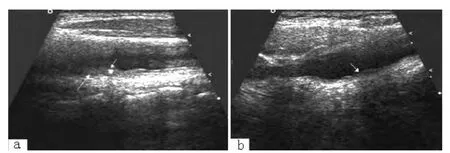
图3 先症者颈动脉超声结果Figure 3 Carotid artery ultrasound of probands
2.2 血脂检测结果 家系1中11例胆固醇增高者TC水平为 (6.98±1.99)mmol/L,24例血脂正常者TC水平为 (3.20±1.02)mmol/L,差异有统计学意义 (t=7.023,P<0.001);胆固醇增高者 LDL-C 水平为(3.02±2.26)mmol/L,血脂正常者 LDL-C 水平为(1.98 ±0.93)mmol/L,差异有统计学意义 (t=3.497,P=0.004)。
家系2中16例胆固醇增高者 TC水平为 (8.12±3.65)mmol/L,32例血脂正常者 TC水平为 (4.37±1.01)mmol/L,差异有统计学意义 (t=4.355,P=0.001);胆固醇增高者 LDL-C水平为 (5.72±3.92)mmol/L,血脂正常者 LDL-C水平为 (2.72±0.62)mmol/L,差异有统计学意义 (t=3.293,P=0.005)。
2.3 基因突变检测结果
2.3.1 apo-B100基因 Q3500R位点突变结果 扩增apo-B100基因3500附近突变位点的片断309 bp,琼脂糖凝胶电泳鉴定 (见图4)。对鉴定后的PCR产物经正反双向核苷酸序列分析 (见图5)及与正常序列比对,均未见患儿apo-B100基因Q3500R位点突变,可排除apo-B100基因与LDL-R结合部位缺陷造成的高胆固醇血症。

图4 琼脂糖凝胶电泳分析apo-B100基因3500附近突变位点的片断Figure 4 Agarose electrophoresis of fragments of apo-B100 3500 mutation sites

图5 apo-B100基因第10708核苷酸序列图Figure 5 The 10708th nucleotide sequence diagram of apo-B100
2.3.2 连锁分析结果 先证者1 LDL-R基因的 LOD值均>2.00,说明该致病基因肯定与LDL-R基因连锁;而PCSK9基因的LOD值0.01~1.11,说明可能连锁,但可能性较小。先证者2的LOD值均<1.00,说明该致病基因与已知突变基因均不连锁,推测可能存在新的致病基因 (见表1)。
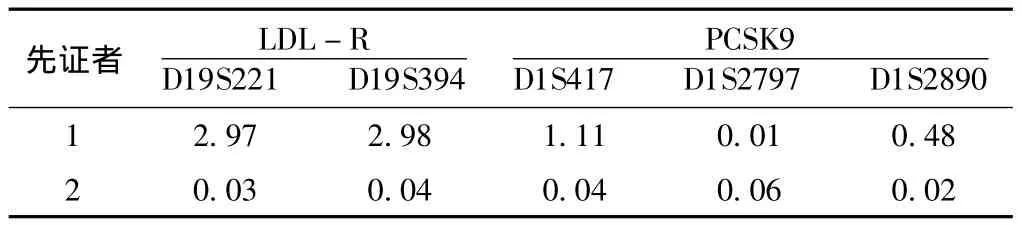
表1 先证者1和先证者2连锁分析定位候选基因的LOD值Table 1 LOD values of linkaged candidate genes in proband 1 and proband 2
2.3.3 核苷酸序列分析结果 LDL-R基因和 PCSK9基因的核苷酸序列分析发现,先证者1 LDL-R基因第2外显子97位点C>T杂合终止突变 (见图6),AAG TAG对应氨基酸由谷氨酰胺变为终止密码子,为Q12X。Q12X在意大利、法国、土耳其人群中有发现,但是在中国该突变非常罕见,由于是终止突变,因此也是致病性突变。先证者1暂未发现PCSK9基因突变。先证者1家系中高脂血症的直系亲属均进行LDL-R基因第2外显子97位点的测序,结果在其父亲、大姑、四姑和五姑中均发现Q12X突变,推测先证者1的致病突变基因来自父系亲属的遗传。先证者2核苷酸序列分析未发现任何已知突变,推测可能存在第4种新的致病基因。

图6 先证者1 LDL-R基因第2外显子核苷酸序列分析结果Figure 6 Nucleotide sequence analysis results of the second exon of LDL-R gene in proband 1
3 讨论
连锁分析使用基因组中与基因突变部位或紧邻的多态性位点作为标记,凡带有该标记的成员可能携带致病基因,从而做出间接诊断。连锁分析作为遗传学初筛的主要方法,已经被美国、丹麦、荷兰等国家广泛使用,国外针对FH的主要致病基因,即LDL-R基因以及新发现的PCSK9基因,采用家系连锁分析的方法,做了大量研究,既为后期测序工作明确了方向,又节省了大量的人力物力[11]。
而我国筛选基因突变的方法普遍滞后于国外发达国家,有技术问题,也有经费问题、成本问题。目前常用的检测FH基因突变的方法有:(1)点突变的检测:主要方法有PCR-单链构象多态性 (SSCP)和变性高效液相色谱分析 (DHPLC)。SSCP是前几年常用的检测FH基因突变的方法,但是SSCP仅对于检测LDL-R基因突变具有高度敏感性,并且仅对LDL-R基因单个碱基置换和某一片段DNA突变位点的筛查有效,不能检测到大片段缺失或重排[12]。DHPLC异源双链的形成取决于高效液相色谱的温度,而色谱温度的选定与野生型DNA融解温度有关,对于温度的控制有一定难度。而越来越多的文献已表明,FH是异质性疾病,LDL-C仅是其中一个常见突变基因类型[13],这显然不适合目前研究现状。(2)大片段缺失或重排的检测:主要包括Southern印迹杂交实验和long-distance PCR。Southern印迹杂交实验过程复杂且涉及的试剂也较多,对DNA的质量要求较高,否则酶切不完全将导致失败,总的来说,Southern印迹杂交实验是一种费时费力的方法。long-distance PCR改善了Southern印迹杂交实验的部分缺点,但其缺点为基因缺失必须在扩增片断所需引物的范围之内,否则缺失的等位基因将不被扩增,从而导致分析结果错误地表现为野生型 (正常)。(3)插入突变的检测:主要采用通用引物荧光定量 PCR(UPQFM-PCR)。但由于其检测基因的单一性和对产物片断的选择性,不提倡将其作为候选基因的初筛方法。(4)拷贝数的检测:荷兰的Eijk-Van Os等[14]所带领的研发小组于2002年发表多重连接探针扩增技术(MLPA)。具有高灵敏度、高特异度、可重复性、简单、高流通量、低成本 (指DNA)。其缺点是:比普通的PCR反应对杂质更敏感 (PCR抑制剂如苯酚残留物等);自行开发探针混合物需要花费相当的时间和精力;与荧光原位杂交技术 (FISH)相比,MLPA不能检测单个细胞DNA拷贝数的变化;只能针对单一基因突变,且费用较高。
近几年,基因芯片发展势头十分迅猛,其中一个重要应用就是基因突变的检测。其原理与经典的核酸分子杂交方法一致,基因芯片在一微小的基片 (硅片、玻片、塑料片等)表面集成了大量的分子识别探针,能够在同一时间内平行分析大量的基因,进行大信息量的筛选与检测分析。但其存在的缺陷也相当明显。首先是成本问题,由于芯片制作的工艺复杂,信号检测也需专门的仪器设备,一般实验室难以承担其高昂的费用;其次在芯片实验技术上还有多个环节尚待提高,如在探针合成方面,如何进一步提高合成效率及芯片的集成程度是研究的焦点。而样品制备的简单化与标准化则是芯片应用进一步普及的前提;再次其操作复杂,结果难以分析,极大限制了基因芯片技术在生命科学研究领域的应用。
以上几种方法均只能作为基因突变筛查的单一着手点,对于FH这类多种致病基因存在的单基因疾病而言,将连锁分析作为候选基因初筛的入手点,具有明显优势。近几年,连锁分析选择的遗传标记迅速发展,由限制性酶切片断到微卫星[15]。由于易于检测,遗传稳定性高、重复性好、省时省力,相比于前几种基因初筛方法更具广泛应用价值。Damgaard等[16]运用最新的微卫星位点 (D1S2890、D19S221、D19S394)设计荧光引物,采用新型毛细管电泳仪,对丹麦20个FH家系共158名成员进行微卫星连锁分析,最终发现多种突变。
本研究所选2个家系均是显性遗传,因此衔接子蛋白(adaptor protein ARH)基因、胆固醇7羟化酶(CYP7A1)基因等隐性遗传的几种致病基因可以直接排除。针对新发现的PCSK9基因和主要的致病基因(LDL-R基因),设计荧光引物,采用 D1S417、D1S2797、D1S2890、D19S221、D19S394等微卫星位点结合新型毛细管电泳仪进行研究,成功建立了中国汉族FH家系PCSK9基因连锁分析的微卫星连锁分析条件,并将现有LDL-R基因微卫星连锁分析的条件适当改变,建立了应用新型毛细管电泳仪分析微卫星峰图的条件,为后期研究做好了准备。
本研究成功发现1例中国罕见突变 Q12X,即LDL-R基因第2外显子97位点C→T杂合终止突变,氨基酸密码子由CAG转变为TAG,编码的谷氨酰胺转变为终止密码子,由于是终止突变,因此也是致病性突变。家系2的测序结果提示可能存在新的未知的致病基因,本课题组下一步将进行全基因组扫描以期发现新的致病基因。
综上所述,连锁分析作为遗传学初筛方法之一,可以很好地应用于FH的基因初筛,并且成功建立了中国汉族FH家系LDL-R基因和PCSK9基因的连锁分析条件,对下一步的基础研究奠定了坚实的基础。
[1] Fellin R,Arca M,Zuliani G,et al.The history of Autosomal Recessive Hypercholesterolemia(ARH).From clinical observations to gene identification [J].Gene,2015,555(1):23-32.
[2] Kolovou G,Vasiliadis I,Gontoras N,et al.Microsomal transfer protein inhibitors,new approach for treatment of familial hypercholesterolemia,review of the literature,original findings and clinical significance [J].Cardiovasc Ther,2015,33(2):71-78.
[3] Ghosh SK,Majumder B,Dutta A.Tuberous xanthoma as a presenting feature offamilialhomozygous hypercholesterolemia with aortic regurgitation [J].J Pediatr,2015,166(1):198.
[4] Goldstein JL,Brown MS.Binding and degradation of low density lipoproteins by cultured human fibroblasts.Comparison of cells from a normalsubjectand from a patientwith homozygous familial hypercholesterolemia[J].J Biol Chem,1974,249(16):5153-5162.
[5] Jones C,Garuti R,Michaely P,et al.Disruption of LDL but not VLDL clearance in autosomal recessive hypercholesterolemia[J].J Clin Invest,2007,117(1):165-174.
[6] Vaverkova H,Soska V,Rosolova H,et al.Czech atherosclerosis society guidelines for the diagnosis and treatment of dyslipidemia in adults[J].Cas Lek Cesk,2007,146(6):Ⅱ-ⅩⅤ.
[7] De Castro-Orós I,Pocoví M,Civeira F.The genetic basis of familial hypercholesterolemia:inheritance,linkage, and mutations [J].Appl Clin Genet,2010,3:53-64.
[8]陈在嘉,徐义枢,孔华宇.临床冠心病学 [M].北京:人民军医出版社,2009:610.
[9] Humphries SE,Whittall RA,Hubbart CS,et al.Genetic causes of familial hypercholesterolaemia in patients in the UK:relation to plasma lipid levels and coronary heart disease risk [J].J Med Genet,2006,43(12):943-949.
[10] No authors listed.Diagnosis,management and prevention of the common dyslipidaemiasin South Africa——clinicalguideline,2000.South African Medical Association and Lipid and Atherosclerosis Society of Southern Africa Working Group[J].S Afr Med J,2002,92(2 Pt 2):164-174,176-178.
[11] Wang X,Li X,Zhang YB,et al.Genome-wide linkage scan of a pedigree with familial hypercholesterolemia suggests susceptibility loci on chromosomes 3q25-26 and 21q22 [J].PLoS One,2011,6(10):e24838.
[12] Komarova TY,Golovina AS,Grudinina NA,et al.New mutations in low-density lipoprotein receptor gene in familial hypercholesterolemia patients from Petrozavodsk [J].Genetika,2013,49(6):773-777.
[13] Page MM,Stefanutti C,Sniderman A,et al.Recent advances in the understanding and care of familial hypercholesterolaemia:significance of the biology and therapeutic regulation of proprotein convertase subtilisin/kexin type 9 [J].Clin Sci(Lond),2015,129(1):63-79.
[14] Eijk-Van Os PG,Schouten JP.Multiplex Ligation-dependent Probe Amplification(MLPA®)for the detection of copy number variation in genomic sequences [J].MethodsMol Biol,2011,688:97-126.
[15] Kudo Y,Nikaido M,Kondo A,et al.A microsatellite-based genetic linkage map and putative sex-determining genomic regions in Lake Victoria cichlids [J].Gene,2015,560(2):156-164.
[16] Damgaard D,Jensen JM,Larsen ML,et al.No genetic linkage or molecular evidence for involvement of the PCSK9,ARH or CYP7A1 genes in the Familial Hypercholesterolemia phenotype in a sample of Danish families without pathogenic mutations in the LDL receptor and apoB genes[J].Atherosclerosis,2004,177(2):415-422.
猜你喜欢
科学导报(2024年19期)2024-04-22 05:53:32
特产研究(2022年6期)2023-01-17 05:05:06
临床输血与检验(2022年3期)2022-06-22 02:52:50
中华养生保健(2020年3期)2020-11-16 00:52:28
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
World Journal of Clinical Cases(2019年6期)2019-04-17 02:25:08
四川动物(2017年4期)2017-07-31 23:54:19
中国卫生标准管理(2015年3期)2016-01-14 03:41:43
重庆医学(2015年12期)2015-03-05 05:52:54
