
聚酰胺-胺-Fe3 O4 复合纳米粒子的制备及细胞毒性研究
2015-07-13 03:12付春华董平轩顾相伶孙汉文宋新峰张彦聪
应用化工 2015年8期
付春华,董平轩,顾相伶,孙汉文,宋新峰,张彦聪
(1.德州学院 医药与护理学院,山东 德州 253023;2.山东省新型药用辅料及缓控释制剂工程实验室,山东 德州 253023)
超顺磁性四氧化三铁纳米粒子具有显著的磁学性能、较低的生物毒性以及良好的生物相容性,这些特点使其在药物磁靶向传输和释放、核磁共振成像、肿瘤热疗、生物传感器与生物探针及酶工程载体等生物医学领域有广阔的应用前景[1-3]。同时,生物体内的巨噬细胞和网状细胞很容易识别IONP,作为转基因载体IONP 可显著提高基因转染效率[4-5]。但是通过物理、化学方法获得的IONP 粒子在应用时存在以下不足:首先,裸磁性粒子在水中分散性差、易聚集沉降,需要表面修饰提高其稳定性;其次,IONP 表面缺乏可利用活性基团[6-8],不易在其表面引入生物活性分子。利用聚合物对IONP 进行包覆是解决上述问题最有效的途径之一。利用高分子本身的特性赋予IONP 良好的生物相容性、分散性及特殊应用目的,我们课题组在前期对IONP 改性、生物学评价的基础上[9-12],用树枝状高分子PAMAM对IONP 进行接枝,并对不同基团封端的复合粒子的细胞毒性进行了初步评价。
1 实验部分
1.1 材料与仪器
FeCl3·6H2O、FeSO4·7H2O、甲醇、甲苯、乙二胺(en)、丙烯酸甲酯(MA)、3-氨基丙基三乙氧基硅烷(3-Aminopropyltriethoxysilan,APTS)均为分析纯;细胞株HepG2,购自北京生化研究所;CCK-8 试剂盒,购自Yeasen 公司。
Zetasizer Nano ZS 马尔文粒度仪;Spectrum GX红外光谱仪;SEM-EDS 扫描电镜-能谱仪;MERLIN Compact 扫描电镜;布鲁克X 射线能谱仪EDS;731型可见分光光度计。
1.2 IONP 的制备
将FeCl3·6H2O 和FeSO4·7H2O 按物质的量之比2∶1 的比例溶解于去离子水,高速搅拌下加入一定量氨水,共沉淀获得超顺磁性、表面带有羟基的IONP。
1.3 PAMAM-IONP 复合粒子的制备
参照文献[12]的工艺路线,IONP 分散于甲苯溶液中超声30 min 后加入APTS,120 ℃回流反应,得到表面氨基化的粒子NH2-IONP(G0)。利用G0表面的氨基与丙烯酸甲酯(MA)在低温下进行迈克尔加成反应后磁分离,获得G0.5 的IONP 粒子。G0.5粒子与过量乙二胺氨解后磁分离获得G1. 0 代粒子。重复迈克尔加成和胺解反应,获得所需代数的PAMAM-IONP 复合粒子。通过控制反应步骤,得到表面为酯键封端和氨基封端不同的粒子。
1.4 可见分光光度计测量复合粒子中聚合物的含量
实验原理是Fe2+与邻二氮菲在pH 为3 ~9 的条件下生成橘红色络合物,该橘红色络合物最大吸收波长为510 nm,可见分光光度计测量[13]。按一定浓度梯度称量IONP 置于容量瓶中,加入盐酸超声波处理将粒子中的铁元素转化为Fe3+和Fe2+。然后加入过量盐酸羟胺将Fe3+转还原为Fe2+后调节pH 值,加入过量邻二氮菲反应规定时间后,测量得到吸光度与Fe3O4浓度的标准曲线。用同样方法处理接枝G5.0 代的复合粒子后测定吸光度,从而间接计算PAMAM 聚合物的质量含量。
1.5 材料细胞毒性试验——MTT 法
实验用纳米粒子经低温间歇灭菌法(60 ℃、10 h,37 ℃、12 h,连续3 次),并用无菌水溶液多次洗涤后,按照不同浓度加入细胞培养基。取对数期的HepG2 细胞在IONP 种类和含量不同的培养基培养24 h,用CCK-8 试剂盒在免疫酶标仪(Multiskan MK3353,USA)上测定492 nm 处的吸光度(OD)值。计算细胞相对增殖率(relative growth rate,RGR),从而判断粒子的细胞毒性。
2 结果与讨论
2.1 IONP 及PAMAM-IONP 复合粒子的特征
使用1.2 节所述共沉淀法制得的IONP 粒子,通过扫描电镜观察,结果见图1。

图1 IONP 的扫描电镜图Fig.1 SEM spectra of IONP
由图1 可知,粒子大小分布均匀、表面相对圆整,直径在10 ~20 nm 之间,符合后期应用对粒子尺寸的要求。少量粒子有团聚现象,使用前超声波处理可得单分散的磁性粒子。
IONP 与PAMAM-IONP 的扫描电镜-能谱(SEMEDS)检测结果见图2。

图2 IONP(1)与PAMAM-IONP(4,G5.0)EDS 图谱Fig.2 EDS spectra of IONP (1)and PAMAM-IONP(4,G5.0)
图2 显示,修饰前后,粒子中Fe、O、C 以及Si 等元素的含量有明显变化。其中,Fe 元素所占质量百分比降低,而O、C 以及Si 等元素的质量百分比增加。为更加清晰的了解这一变化趋势,将各元素相应的含量计算并列于表1 中。表中数据进一步表明,随着修饰代数的增加,C 元素的含量逐渐增加。该结果初步表明:PAMAM 成功的接枝IONP,获得目标产物复合粒子。
此外,表1 中也给出了所得粒子的水相粒径和电位结果。与SEM 观察所得粒径不同,马尔文粒度仪测试IONP 粒径结果为696.2 nm。究其原因,我们认为,由于裸IONP 粒子在水溶液中稳定性差、易聚沉,因此采用激光散射测试粒径出现了较大误差。粒子接枝修饰后,由于在水溶液中稳定性增强、分散性得以改善,马尔文粒度仪测试亦较为真实的反映了其粒径。从粒子电位的测试结果来看,使用PAMAM 对IONP 表面修饰后,其电位从- 4. 28 mV 变化 为-23.30 mV,证明修饰后的粒子得以在水相稳定性增强,进一步证明PAMAM 成功的接枝IONP。

表1 不同粒子的粒径、电位及EDS 元素检测结果Table 1 The test data of particle size,potential and element content
2.2 红外检测结果
使用红外光谱仪对不同粒子进行测试,结果见图3。
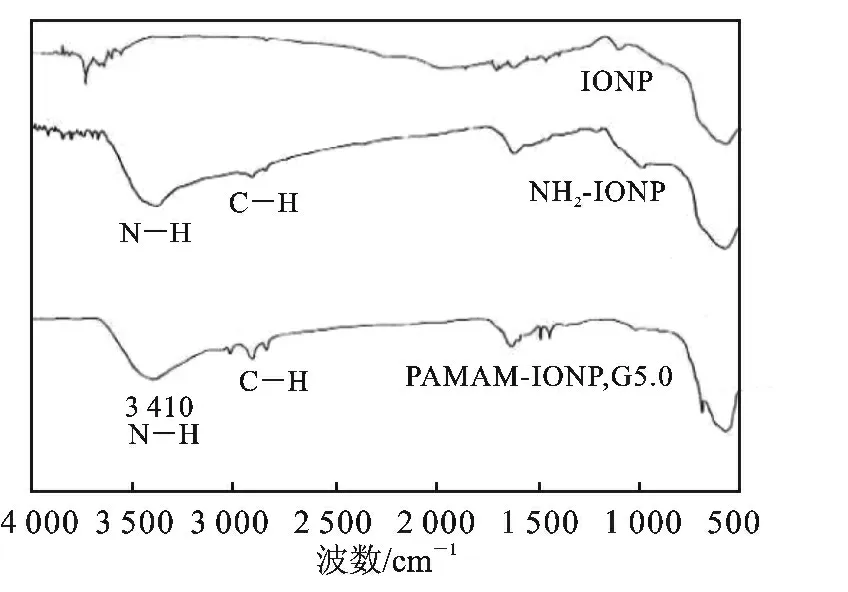
图3 不同粒子的红外图谱Fig.3 FTIR spectra of different particles
由图3 可知,与裸磁粒子的红外谱图相比,APTS 氨 基 化 的 IONP 和 PAMAM-IONP 在3 400 cm-1附近出现显著的吸收,对应于N—H 特征吸收峰,2 800 ~2 900 cm-1处的吸收对应于C—H的特征吸收峰。上述结果进一步表明,使用1.3 节所述方法可成功制备PAMAM-IONP 复合粒子。
2.3 壳层聚合物质量含量测试结果
为进一步了解IONP 表面包覆的PAMAM 含量,用分光光度法测试了不同粒子的吸光度。IONP 粒子浓度与吸光度A 的标准曲线见图4。
标准曲线的相关系数达到了0.991,该曲线可用于粒子浓度的标定。相关测试结果表明,接枝5.0 代的PAMAM-IONP 外层聚合物的平均质量含量为(13.6 ±0.3)%,实验结果重现性较好,复合定量测定要求。
2.4 细胞毒性实验结果
将裸IONP、氨基封端复合粒子(PAMAM-IONP,G5. 0)和 酯 键 封 端 的 粒 子(MA-PAMAM-IONP,G5.5)分别以5,10,50,100,200,500 μg/mL 浓度梯度的培养液与HepG2 细胞共培养结果见图5。

图4 Fe3O4粒子浓度与吸光度A 的标准曲线Fig.4 The standard curve of Fe3O4 concentration and absorption
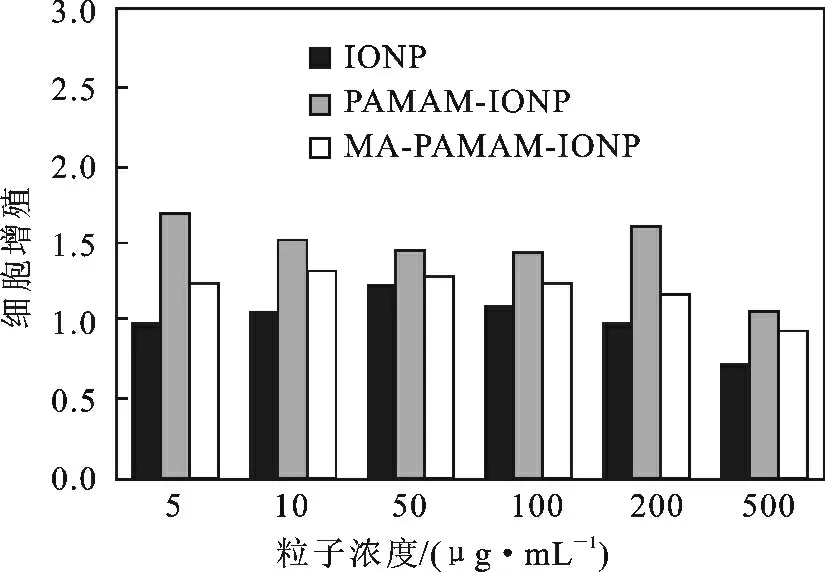
图5 不同类型、不同浓度的粒子对细胞增殖的影响Fig.5 Effect of different particles on HepG2 cell proliferation
由图5 可知,同一种磁性粒子的浓度低于200 μg/mL时,浓度对细胞的增殖性影响没有明显差异,但是当粒子浓度高至500 μg/mL 时,对细胞增殖的抑制性明显增加。比较3 种粒子对细胞培养结果可以发现,浓度相同时,裸SIONP 细胞增殖数最小,而氨基封端的复合粒子细胞增殖数最高,表明氨基端的粒子细胞毒性最小,细胞安全性最佳,上述结果与文献[14]报道一致,实现了材料表面改性修饰的目的。
3 结论
目前IONP 的应用主要集中在生命科学领域,因此对其修饰改性材料的生物安全性评价是能否进行后期研究的关键环节,而细胞毒性评价是生物安全评价的重要组成部分。本研究结果表明,采用树枝状高分子PAMAM 修饰的IONP 不仅改善了粒子在水溶液中的分散性和稳定性,其细胞毒性也有明显下降,且细胞毒性与粒子表面的基团的种类有关。因此后期根据不同的用途设计合理的分子结构提供了依据。
[1] Jeong U,Teng X W,Wang Y,et al. Magnetic nanoparticles:preparation,physical properties,and applications in biomedicine[J].Advance Materials,2007,19:33-60.
[2] Weinstein J S,Varallyay C G,Dosa E,et al. Superparamagnetic iron oxide nanoparticles:diagnostic magnetic resonance imaging and potential therapeutic applications in neurooncology and central nervous system inflammatory pathologies[J].Journal of Cerebral Blood Flow & Metabolism,2010,30:15-35.
[3] Nakai G,Matsuki M,Harada T,et al.Evaluation of axillary lymph nodes by diffusion-weighted MRI using ultrasmall superparamagnetic iron oxide in patients with breast cancer:Initial clinical experience[J].Journal of Magnetic Resonance Imaging,2010,30:15-35.
[4] Pankhurst Q A,Connolly J,Jones S K,et al.Applications of magnetic nanoparticles in biomedicine[J]. Journal of Physics D:Applied Physics,2003,36:167-181.
[5] Akbarzadeh A,Samiei M,Davaran S.Magnetic nanoparticles:preparation,physical properties,and applications in biomedicine[J]. Nanoscale Research Letters,2012,7:144-152.
[6] Issa B,Obaidat I M,Albiss B A,et al.Magnetic nanoparticles:surface effects and properties related to biomedicine applications[J]. International Journal of Molecular Sciences,2013,14:21266-21305.
[7] Kostiantyn Turcheniuk,Arkadii V Tarasevych,Valeriy P Kukhar,et al.Recent advances in surface chemistry strategies for the fabrication of functional iron oxide based magnetic nanoparticles [J]. Nanoscale,2013,5:10729-10752.
[8] Ruiz A,Morais P C,Azevedo R B,et al.Magnetic nanoparticles coated with dimercaptosuccinic acid:development,characterization,and application in biomedicine[J]. Journal Nanoparticals Research,2014,16:2589-2608.
[9] Sun H W,Zhu X J,Zhang L Y,et al.PDEA-coated magnetic nanoparticles for gene delivery to Hep G2 cells[J].Biotechnology and Bioprocess Engineering,2013,18:648-654.
[10]孙汉文,张连营,朱新军,等.PEGMA 磁性纳米凝胶的光化学原位合成、表征及载药性能研究[J].中国科学(B 辑:化学),2008,11:999-1005.
[11]孟繁宗,孙汉文,张连营,等. 荧光磁性微球Fe3O4@PMAA-Dy 的制备和表征[J]. 材料导报,2009(6):133-136.
[12]Wang J Z,Zhao G H,Li Y F.Reversible immobilization of glucoamylase onto magnetic chitosan nanocarriers[J].Appl Microbiol Biotechnol,2013(97):681-692.
[13]Zhang C,Jiang M G,Huang W,et al. Determination of iron ions in cooling water by spectrophotometry[J]. Journal of Salt and Chemical Industry,2013(11):45-47.
[14]Rasala R M,Janorkarc A V,Hirta D E.Poly(lactic acid)modifications[J]. Progress in Polymer Science,2010(35):338-356.
猜你喜欢
当代水产(2021年6期)2021-08-13
合成树脂及塑料(2020年6期)2020-12-29
小哥白尼(野生动物)(2019年5期)2019-08-27
中成药(2018年11期)2018-11-24
智富时代(2018年7期)2018-09-03
智富时代(2018年7期)2018-09-03
中国塑料(2016年3期)2016-06-15
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
中国塑料(2015年1期)2015-10-14
中国生化药物杂志(2015年4期)2015-07-07
