
下调人脐带间充质干细胞Mbd3基因shRNA慢病毒的构建及鉴定
2015-06-09 14:24闫姗姗孙茂盛解裕萍李鸿钧
生物学杂志 2015年3期
闫姗姗,周 艳,李 溪,张 磊,孙茂盛,解裕萍,李鸿钧
(1.中国医学科学院北京协和医学院医学生物学研究所云南省重大传染病疫苗研发重点实验室,昆明650118;2.昆明医科大学附属第一医院骨科,昆明650032)
下调人脐带间充质干细胞Mbd3基因shRNA慢病毒的构建及鉴定
闫姗姗1,周 艳1,李 溪2,张 磊1,孙茂盛1,解裕萍1,李鸿钧1
(1.中国医学科学院北京协和医学院医学生物学研究所云南省重大传染病疫苗研发重点实验室,昆明650118;2.昆明医科大学附属第一医院骨科,昆明650032)
研究构建靶向抑制人Mbd3表达的shRNA慢病毒颗粒,感染人脐带间充质干细胞并用嘌呤霉素筛选获得Mbd3稳定下调的细胞系,为进一步探讨Mbd3在诱导型多潜能干细胞形成中的作用提供实验基础。研究根据Gene Bank中公布的Mbd3基因序列设计特异性siRNA干扰序列并合成可产生shRNA的双链DNA,经双酶切后克隆至pLVshRNA-EGFP(2A)Puro载体上构建慢病毒重组质粒,转化DH5α大肠杆菌PCR检测筛选阳性质粒,利用HEK293Ta包装产生含有Mbd3shRNA的慢病毒颗粒。通过RT-PCR和Western blot检测慢病毒感染人脐带间充质干细胞(hUC-MSC)后Mbd3的转录和蛋白表达情况,进一步检测重组慢病毒对Mbd3的沉默效果并筛选出最佳抑制效果的shRNA慢病毒载体。结果显示成功构建并筛选出下调Mbd3的最佳慢病毒干扰载体,慢病毒感染人脐带间充质干细胞48h后于荧光显微镜下可见绿色荧光,经嘌呤霉素筛选9 d后,RT-PCR检测Mbd3表达显著降低,Western blot检测Mbd3蛋白表达明显减少。说明构建的shRNA重组慢病毒包装成功,具有较高的感染活性,可有效抑制人脐带间充质干细胞Mbd3的表达,为后续研究脐带间充质干细胞跨胚层分化打下前期研究基础。
Mbd3基因;基因沉默;慢病毒载体;RNA干扰
Mbd3是甲基化CpG结合蛋白家族中的主要成员,能与甲基化CpG二核苷酸特异性结合并相互作用,也是核小体去乙酰化复合体(NuRD)的核心组成部分,通过DNA甲基化和组蛋白去乙酰化进行表观遗传修饰[1-3]。但Mbd3并不像其他MBD蛋白一样,不与甲基化的CpG结合,而是直接与NuRD的亚单位Chd4结合[4]。Mbd3广泛存在于哺乳动物、昆虫、线虫、植物的几乎所有细胞中,这种现象十分罕见,也因此引起了研究工作者对Mbd3的关注。Mbd3对早期的细胞发育,维持胚胎干细胞的多能性和多能细胞的发生发展起着重要的作用[5-6]。研究表明,Mbd3可以通过抑制Cdx2的表达从而抑制鼠胚胎干细胞向滋养外胚层分化,对维持鼠胚胎干细胞的全能性起到重要的表观调控作用[7-8]。Keisuke等[1]发现抑制Mbd3的表达能够阻止内细胞团向外胚层的分化并进一步推测Mbd3/NuRD对多能细胞的产生起着关键作用。2013年Rais等[9]人发现过表达Oct4,Sox2,Klf4和Myc的同时抑制Mbd3的表达可快速高效地将体细胞诱导成iPSC,有力的证明了Kaji等的推测[1]。在前期研究中,我们能够将间充质干细胞向神经元样细胞诱导分化,但分化效率不高。因此,我们提出假设,直接下调间充质干细胞Mbd3的表达是否同样可提高间充质干细胞的多向分化能力,提高细胞的跨胚层分化潜能,为进一步研究iPSC的形成奠定基础。本实验旨在通过构建Mbd3 shRNA慢病毒载体并转染人脐带间充质细胞,筛选稳定下调Mbd3表达的细胞株,为将来研究脐带间充质干细胞跨胚层分化及诱导为神经细胞的优化条件打下前期研究基础。
1 材料和方法
1.1材料
人脐带间充质干细胞(hUC-MSC)从新生儿脐带组织中分离得到,HEK293Ta细胞为本实验室保存,DMEM高糖培养基(CORNING),MEM-α培养基(Hy-Clone),Opti-MEM(Gibco),胎牛血清(Biological Industries),EndoFectin-Lenti转染试剂购自广州复能基因有限公司,pLVshRNA-EGFP(2A)Puro载体购自北京英茂盛业生物技术有限公司,Eco RI和Bam HI限制性内切酶(TaKaRa),T4 DNA连接酶(TaKaRa),质粒小提试剂盒(Omega),DH5α大肠杆菌本实验室保存。SDS-PAGE凝胶制备试剂盒(康为世纪生物技术有限公司),PVDF膜(M illipore),兔抗人Mbd3一抗(Cell Singnaling),HRP标记的山羊抗兔IgG(KLP)。高纯总RNA提取试剂盒(百泰克),RNA逆转录试剂盒(Thermo),Real-time PCR试剂(BIO-RAD)。
1.2方法
1.2.1细胞的培养
取健康新生儿脐带剪成2~3 cm,用含1%双抗的PBS溶液充分洗涤残留在脐带表面的血渍,剔除脐带中的脐静脉、脐动脉、华尔通氏胶(Wharton’s Jelly,WJ),将脐带最外侧的组织剪碎至1mm×1mm×1 mm大小,接种于T-25 cm2的细胞培养瓶中,倒置放入37℃、5%CO2培养箱中培养1~2 h,待组织块贴壁牢固后加入含有15%胎牛血清,1%双抗,1%谷氨酰胺的α-MEM完全培养基,于37℃、5%CO2培养箱中培养。培养期间于显微镜下观察细胞生长情况,待细胞融合达到60%~70%时,即可去除组织块继续培养,当细胞密度达90%左右,用0.125%胰酶消化,1∶2传代培养。293Ta细胞用含10%胎牛血清、1%双抗的DMEM高糖培养基,37℃,5%CO2细胞培养箱培养。观察细胞生长状态,待细胞密度达90%时用0.25%胰酶消化,1∶2或1∶3传代培养。
1.2.2 Mbd3特异性靶序列的设计及慢病毒载体的构建
Mbd3的特异性靶序列由北京英茂盛业生物技术有限公司设计并合成含干扰序列的双链DNA oligo 4条。其特异性靶序列分别为:Mbd3 shRNA1-CGGTG ACCAAGATTACCAA,Mbd3 shRNA2-CATTGACCT TAGGCCCATA,Mbd3 shRNA3-CTGTCAGAGTCAA AGCACA,Mbd3 shRNA control-ATCGACTAGCCACT TAGAC。Eco RI和Bam HI对双链DNA oligo和载体进行双酶切后用T4 DNA连接酶16℃链接过夜构建pLVMbd3 shRNA-EGFP-2A环状质粒。将连接产物转化DH5α大肠杆菌感受态细胞,挑取单菌落于LB液体培养基中37℃,240 r/m in培养过夜,以提取慢病毒重组质粒为模板进行PCR,筛选阳性克隆,PCR引物为:U6F-GACTATCATATGCTTACCGTAAVT和 U6RGGTGGATGTGGAATGTGTG,反应条件:95℃预变性2m in;95℃30 s,56℃30 s,72℃30 s,共35个循环;72℃10m in。并将筛选到的阳性质粒送去大连宝生物公司测序。
1.2.3慢病毒的包装
制备慢病毒穿梭质粒pLVMbd3 shRNA-EGFP-2A及其慢病毒包装辅助质粒PH1和PH2,3种质粒共转染HEK293Ta细胞后,在HEK293Ta细胞中PH1和PH2表达产生病毒颗粒所需的病毒蛋白并与穿梭质粒组装成有复制缺陷的慢病毒颗粒。将生长状态良好的HEK 293Ta细胞传至直径为10 cm的双碟中,所用培养基为含10%FBS和1%双抗的DMEM高糖培养基,待细胞密度达60%~70%时,换成含10%热灭活FBS的DMEM高糖培养基,pLVMbd3 shRNA-EGFP-2A、PH1、PH2 3种质粒按一定比例与Opti-MEM混合,EndoFectin-Lenti转染试剂与Opti-MEM混合,静置5 min后将质粒与转染试剂混合,逐滴加入细胞中,于37℃,5%CO2细胞培养箱中孵育过夜,更换培养基为含5%热灭活FBS和1%双抗的DMEM高糖培养基,分别于48 h和72 h收集含有病毒颗粒的细胞上清液。1000 r/m im离心1m in去除细胞碎片,取上清液于-80℃保存,供后续实验使用。
1.2.4慢病毒滴度测定
采用Real-time PCR法测病毒滴度。检测前一天将HEK293Ta细胞传至24孔板中,次日用含10%FBS的DMEM培养基10倍梯度稀释病毒液,取100μL稀释好的病毒液加入细胞孔中于37℃5%CO2培养箱中培养,4 d后抽提细胞总RNA,逆转录试剂盒将总RNA逆转录成cDNA,PCR扩增程序为25℃5min,42℃60 m in,70℃5m in。以cDNA为模板Real time PCR检测EGFP基因表达情况,上游引物:TGCTTCAGCCGCTACCC,下游引物:AGTTCACCTTGATGCCGTTC。PCR扩增条件为:95℃预变性3 m in;95℃20 s,56℃20 s,72℃20 s,82℃5 s,共45个循环。溶解曲线65℃~95℃,每一步增加0.5℃,保持5 s。
1.2.5慢病度颗粒感染hUC-MSC及稳定细胞株的筛选
将生长状态良好的hUC-MSC细胞传至T-25细胞培养瓶中,待细胞密度达70%时,将细胞液换成含5% FBS,1%双抗,5μg/m L ploybrene的DMEM高糖培养基并加入收获的病毒液,第2天换成含5%FBS,1%双抗的DMEM高糖培养基,48 h后于荧光显微镜下观察绿色荧光蛋白表达情况,4 d后加嘌呤霉素对细胞进行筛选,筛选9 d左右后提取细胞总蛋白及总RNA。
1.2.6 Real-time PCR检测Mbd3转录情况
从稳定转染了Mbd3 shRNA慢病毒的hUC-MSC细胞中提取细胞总RNA并逆转录成cDNA,以cDNA为模板Real-time PCR检测Mbd3表达情况,以β-actin为内参,所用引物序列如下:β-actin up:ACGTT GACATCCGTAAAGAC,β-actin down:GAAGGTACA GTGAGGC,Mbd3 up:TCAAGAGCGACCCGCAGA A,Mbd3 down:TGTCCCGTGATGGGCATG。PCR扩增条件为:95℃预变性3m in;95℃20 s,56℃20 s,72℃20 s, 82℃5 s,共45个循环。溶解曲线65℃~95℃,每一步增加0.5℃,保持5 s。
1.2.7 Western blot检测Mbd3蛋白表达情况
从稳定转染了Mbd3 shRNA慢病毒的hUC-MSC细胞中提取细胞总蛋白并用Lorrery法测定所提蛋白浓度,使各样品蛋白浓度一致进行SDS-PAGE聚丙烯酰胺凝胶电泳,通过电转将凝胶上的蛋白转至PVDF膜上,封闭液封闭2 h后孵育兔抗人Mbd3一抗(1∶1000稀释),PBST洗涤PVDF膜后孵育HRP标记的山羊抗兔二抗(1∶2000稀释),PBST洗涤后胶片曝光观察结果。
2 结果
2.1 hUC-MSCs的分离及培养
剪碎的脐带组织块贴壁后7 d左右可见贴壁生长的成纤维样细胞出现,细胞呈克隆样生长。普通光学显微镜下观察细胞形态,细胞核较大,细胞呈长梭形、短棒状或扁平形,折光性好,核仁清晰可见,培养至10 d左右,细胞量明显增多,细胞形态变为均一的纺锤形,呈旋涡状生长(图1A)。原代细胞1∶2传代后,细胞分布均匀,贴壁生长,细胞核较大,有的可清晰看到核仁,形态呈长梭形,平行生长或漩涡生长(图1B)。3~4 d细胞密度达90%,可进行传代,体外至少可以传代7次。
2.2 Mbd3 shRNA慢病毒载体的构建及鉴定
以重组慢病毒载体pLVMbd3 shRNA-EGFP-2A和空慢病毒载体为模板进行RCR并电泳检测PCR产物大小,结果显示空载体可扩增得到212 bp的DNA片段,pLVMbd3 shRNA-EGFP-2A可得到260 bp的DNA片段(图2)。测序结果也证明重组慢病毒载体pLVMbd3 shRNA-EGFP-2A构建成功。
2.3 Mbd3 shRNA重组慢病度的包装及滴度测定
EndoFectin-Lenti转染试剂介导pLVMbd3—shRNAEGFP-2A、PH1和PH2转染HEK293Ta细胞,培养48 h后在荧光显微镜下观察,可见HEK293Ta表达绿色荧光(图3A和3B),说明pLVM bd3—shRNA-EGFP-2A成功转染HEK293Ta细胞,并用收获的病毒液感染HEK293Ta细胞48 h后荧光显微镜观察到绿色荧光,未被慢病毒感染的细胞不表达绿色荧光蛋白(图3C、D、E、F),说明成功包装出具有感染活性的慢病毒颗粒。Real-timePCR检测病毒滴度为2×106TU/m L(图4)。

图1 分离得到的原代hUC-MSCs(A)与传代的hUC-MSCs(B)Fig 1 The cultureof primary hUC-MSCs(A) and sub-culturehUC-MSCs(B)
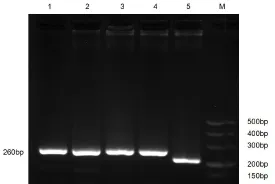
图2 构建的相关载体PCR鉴定结果Fig 2 The resultof PCR related vector1、2、3和4—以重组慢病毒载体pLVMbd3 shRNA-EGFP-2A为模板;5—以慢病毒空载体为模板;M-DL500DNAMarker。
2.4 Mbd3 shRNA重组慢病度感染hUC-MSC及稳定细胞株的筛选
以2×104TU/m L的Mbd3 shRNA重组慢病度感染hUC-MSC细胞,培养48 h后与荧光显微镜下可观察到绿色荧光,而对照组细胞不表达绿色荧光蛋白(图5),感染4 d后加入5μg/m L的嘌呤霉素对细胞进行筛选,筛选过程中可观察到未被重组慢病度感染的hUCMSC细胞逐渐死亡,筛选9 d后可对细胞进行传代。
2.5 Real-time PCR检测慢病毒感染的hUC-MSCs中Mbd3转录情况
提取4种Mbd3 shRNA重组慢病度(Mbd3 shRNA1、Mbd3 shRNA2、MbdshRNA control)感染的hUCMSC细胞总RNA,以逆转录的cDNA为模板在mRNA水平检测Mbd3转录情况。与对照组相比较Mbd3 shRNA1、Mbd3 shRNA2和Mbd3 shRNA3对Mbd3的表达均有抑制效果,其中Mbd3 shRNA1和Mbd3 shRNA3的抑制效果更好(图6)。
2.6 Western blot检测慢病毒感染的hUC-MSCs中Mbd3蛋白表达情况
提取4种Mbd3 shRNA重组慢病度(Mbd3 shRNA1、Mbd3 shRNA2、MbdshRNA control)感染的hUC-MSC细胞总蛋白。Westernblot检测结果表明:3个实验组与对照组相比较Mbd3蛋白表达均有减少,其中Mbd3 shRNA1和Mbd3 shRNA3下调效果更好,Mbd3 shRNA3可以达到完全抑制的作用(图7)。

图4 Rea l-tim e PCR法测定慢病毒滴度Fig 4 The concentration determ ination of lentivirusby Real-time PCR
3 讨论

图5 慢病毒感染hUC-MSCs绿色荧光蛋白表达情况Fig 5 Theexpression of EGFPin hUC-MSCsafter infected by lentivirusA、B—重组慢病毒pLVMbd3 shRNA-EGFP-2A感染,hUC-MSCs绿色荧光蛋白表达情况;C、D—hUC-MSCs对照组绿色荧光蛋白表达情况
细胞替代治疗是指重建受损的组织结构,恢复其正常功能从而达到治疗疾病的目的。目前已经证明多种疾病适用于细胞替代性治疗,如癌症、糖尿病、神经退行性疾病、心血管疾病、视网膜变性疾病、青光眼等。但目前对用于细胞替代性治疗的种子细胞的研究主要局限在胚胎干细胞,虽然不乏许多成功的案例,但由于这些细胞的获取方式特殊和应用时的不可预测性,使它不可避免的带来一系列伦理问题,细胞来源受到很大的争议,引起了卫生系统、医疗机构、患者、研究者及投资者之间的利益冲突,使细胞替代治疗广泛应用于临床受到极大的限制。随着2006年诱导性多潜能干细胞(iPSC)的发现,给细胞替代性治疗的广泛应用带来了曙光,也引起人们对iPSC的研究热潮。
Mbd3作为NuRD的核心组成部分自1998年被发现以来,经过大量研究证实,Mbd3的生化性质并不像他名称所描述的那样有可与甲基化的CpG岛链接结构域,功能上也不像MBD家族蛋白一样募集成NuRD后将DNA甲基化,包含Mbd3的NuRD复合物是通过序列特异性连接蛋白直接与靶基因结合定位染色质修饰部位使组蛋白H3和H4氨基末端去乙酰化,从而沉默基因的转录[10]。为了确定Mbd3的功能,Kaji等[11]在缺乏细胞分化抑制因子LIF的条件下同时培养ES细胞和Mbd3缺陷的ES细胞发现Mbd3缺陷的胚胎干细胞可以长期保持未分化状态(长达30 d)并表达多能基因Oct4和Rex1,而野生型胚胎干细胞很快分化并下调了Oct4和Rex1的表达。进一步实验发现Mbd3缺陷的ES细胞破坏了胚胎的正常发育和形态发生,说明抑制Mbd3的表达可以限制干细胞的分化,提高细胞自我更新能力。近些年的大量研究也表明Mbd3调控基因的表达,抑制干细胞的分化,维持干细胞的多能性,促进体细胞及多能干细胞向iPSC分化,重编程体细胞基因的表达[12-13]。Mbd3表达于发育每个阶段的每一个细胞中,只在受孕后的头3天不表达,而受精卵正是在这3天中开始分裂,可供给机体所需的所有细胞类型。从第4天开始分化启动,并且细胞开始丧失它们的多能性,也就是在这时Mbd3蛋白才第一次出现,这一现象十分罕见,其对于产生医用iPSCs具有重大的意义。

图6 Rea l-tim e PCR检测慢病毒重组质粒对hUC-MSCs中MBD3基因表达的抑制效果Fig 6 The Real-time PCR analysis resultof Mbd3 in hUC-MSCD—重组慢病毒pLV Mbd3shRNA-EGFP-2A感染hUC-MSCs 9 d后Mbd3 mRNA水平。Huc1:1:pLV Mbd3shRNA 1-EGFP-2A;huc2:pLV Mbd3shRNA2-EGFP-2A;huc3:pLV Mbd3shRNA 3-EGFP-2A;huc control:pLV Mbd3shRNA control-EGFP-2A。
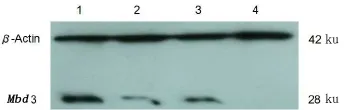
图7 Western blot检测慢病毒重组质粒对hUC-MSCs中Mbd3蛋白表达情况Fig 7 TheWestern blotanalysisof Mbd3 in hUC-MSC1—pLVMbd3shRNA control-EGFP-2A;2—pLVMbd3shRNA 1-EGFP-2A;3—pLVMbd3shRNA2-EGFP-2A;4—pLVMbd3shRNA 3-EGFP-2A。
RNA干扰是指内源性或外源性双链RNA(doublestranded RNA,dsRNA)与细胞内同源序列的mRNA特异性结合,导致mRNA降解抑制相应蛋白的表达。广泛用于探索基因功能和传染性疾病及恶性肿瘤的基因治疗领域。RNAi技术具有高特异、高效率、可遗传、操作简单、可同时进行多基因研究等优点。目前常用的方法主要有脂质体法、阳离子聚合物、电穿孔法、慢病毒介导发等。各种方法的干扰效率及作用时间长短各有不同,但因慢病毒具有感染效率高、感染细胞范围广,既能感染分裂活跃的细胞,又能感染非分裂或分裂缓慢的细胞、可将外源基因整合到宿主染色体上达到稳定持久的表达效果、不在宿主细胞繁殖,不会引起宿主细胞死亡、携带外源基因较大等优点,所以被作为体内体外基因传递的有效工具[14-15]。
本实验首先成功构建pLVMbdshRNA-EGFP(2A)重组慢病毒载体,并应用HEK293Ta细胞包装产生具有感染活性的慢病毒颗粒。因慢病毒载体携带有EGFP基因和Puromycin基因,所以可通过荧光显微镜观察绿色荧光蛋白表达情况确定慢病毒感染情况并粗略判断其感染效率,通过嘌呤霉素筛选稳定转染的细胞系。TR-PCR结果显示,与对照组相比pLVMbd3 shRNA1-EGFP(2A)和pLVMbd3 shRNA2-EGFP(2A)两组Mbd3的mRNA水平均明显下降。Western blot检测结 果 也 表 明 pLVMbd3 shRNA1-EGFP(2A)和pLVMbd3shRNA3-EGFP(2A)两组Mbd3蛋白的表达明显下降,与RT-PCR结果一致。表明慢病毒介导的shRNA能高效稳定沉默Mbd3的表达。
综上所述,我们成功构建了pLVMbd3shRNAEGFP(2A)重组慢病毒载体,包装产生了具有高感染活性的慢病毒颗粒,建立了稳定下调Mbd3表达的hUCMSC细胞系。为进一步研究Mbd3对iPS产生的作用以及为将来成体干细胞直接跨胚层分化研究打下了前期基础。
[1]Keisuke K,Nicholsa J,Hendrich B.Mbd3 a component of the NuRD co-repressor complex,is required for development of pluripotentcells[J].Development,2007,134:1123-1132.
[2]Zhang Y,Ng H H,Erdjument-Bromage H,et al.Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection w ith DNA methylation[J].Genes Development, 1999,13:1924-1935.
[3]Xue Y T,Wong JM,Moreno G Y,etal.NURD,a novel complex w ith both ATP-dependent chromatin-remodeling and histone deacetylaseactivities[J].Cell Press,1998(2):851-861.
[4]Zhu D M,Fang JS,Zhang J.Mbd3,a Componentof NuRD/M i-2 complex,helps maintain pluripotency of mouse embryonic stem cells by repressing trophectoderm differentiation[J].PLoSONE, 2009,4(11):e7684.
[5]Bowen N J,Fujita N,Kajita M,et al.M i-2/NuRD:multiple complexes for many purposes[J].Biochimicaet Biphysica Acta, 2004,1677:52-57.
[6]Reynolds N,Latos P,Hynes-A llen A,et al.NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage comm itment[J].Cell Stem Cell,2012,10: 583-594.
[7]Yildirim O,Li R W,Hung J H,et al.Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosinemarked genes in embryonic stem cells[J].Cell,2011,147:1498-1510.
[8]Brumbaugh J,Hochedlinger K.Removing reprogramm ing roadblocks:Mbd3 depletionallows deterministic iPSC generation[J]. CellStem Cell,2013,13:379.
[9]Rais Y,Zviran A,Geula S,et al.Deterministic direct reprogramm ing of somatic cells to pluripotency[J].Nature,2013,502 (7469):65-70.
[10]Hendrich B,Bird A.Identification and characterization of a family ofmammalian methyl-CpG binding proteins[J].Molecularand Cellular Biology,1998,18(11):6538-6547.
[11]Crook JM,Dunn N R,Colman A.Repressed by a NuRD[J]. Nature CellBiology,2006,8:212-214.
[12]Reynolds N,O’Shaughnessy A,Hendrich B.Transcriptional repressors:multifaceted regulators of gene expression[J]. Development,2013,140:505-512.
[13]dos Santos R L,Tosti L,Radzisheuskaya A,et al.MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner[J].CellStem Cell,2014,15:102-110.
[14]WangW,Zhu H,Zhang H,etal.Targeting PPM 1D by lentivirusmediated RNA interference inhibits the tumorigenicity of bladder cancer cells[J].Brazilian Journal of Medical and Biological Research,2014,47(12):1044-1049.
[15]Chen F L,Lin P F,Li X,et al.Construction and expression of lentiviral vectors encoding recombinantmouse CREBZF in NIH 3T3 cells[J].Plasmid,2014,76:24-31.
Construction and appraisalof lentiviralshRNA vector silencehuman M bd3 gene expressing
YAN Shan-shan1,ZHOU Yan1,LIXi2,ZHANG Lei1,SUNMao-sheng1, XIEYu-ping1,LIHong-jun1
(1.Institute ofMedicalBiology,Chinese Academy ofMedicalSciences,Peking Union MedicalCollege, Yunnan Key Laboratory of Vaccine Research&Developmenton Severe InfectiousDisease,Kunm ing 650118;2.FirstA ffiliated Hospitalof Kunm ing MedicalUniversity,Kunm ing 650032,China)
In this study,we constructed a shRNA lentivirus particle,it is able to targetedly suppress Mbd3 expression and infecthuman umbilical cordmesenchymal stem cells(hUC-MSCs)lines screened by puromycin in which Mbd3 is down regulated stably.Specific siRNA was designed and dsDNA that could generate shRNA was synthesized based on the gene sequence of Mbd3.After double digestion,pLVshRNA-EGFP(2A)Puro vector was used to construct recombination lentivirus plasm id and transformed into E.coli DH5αsubsequently.The positive plasm id was screenedby PCR.Packaged lentivirus particle contained Mbd3 shRNA by HEK293Ta.Then hUC-MSCswere infected w ith this lentivirusand the effectof recombination lentiviruson theMbd3 gene silencewas detected through analysisofmRNA transcription level and protein level.The results showed that the lentivirus vectorwhich could interfere inMbd3 gene expression efficaciously was constructed and screened.Green fluorescence was observed through fluorescence microscope 48 h after hUC-MSCs were infected w ith this lentivirus and the expressions of Mbd3 gene and Mbd3 protein were significantly decreased through the detection by RT-PCR and Western blot 9 days after screening by puromycin.
Mbd3 gene;gene silencing;lentiviralvector;RNA interference
Q782;R556.5
A
2095-1736(2015)03-0015-06
10.3969/j.issn.2095-1736.2015.03.015
2014-11-11;
2014-11-21
资助基金:云南省重点新产品开发计划(2012AE001);协和青年基金和中央高校基本科研业务费专项资金(33320140082)
闫姗姗,硕士,研究方向:生物化学与分子生物学相关研究;
李鸿钧,研究员,长期从事干细胞及分子生物学相关研究,E-mail:lihj6912@hotmail.com。
猜你喜欢
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年5期)2021-07-22
现代临床医学(2021年2期)2021-03-29
世界科学技术-中医药现代化(2021年10期)2021-03-02
中国生殖健康(2020年2期)2021-01-18
健康博览(2019年10期)2019-12-02
中国生殖健康(2018年2期)2018-11-06
食品科学(2018年10期)2018-05-23
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
