
IL-10抑制TGF-β1诱导的大鼠心脏成纤维细胞增殖及表型转化
2015-06-05 05:10郝燕捷韩晓宁丁文惠
基础医学与临床 2015年9期
郝燕捷,陈 莹,薛 林,韩晓宁,丁文惠*
(北京大学第一医院1.心内科;2.风湿免疫科,北京 100034)
IL-10抑制TGF-β1诱导的大鼠心脏成纤维细胞增殖及表型转化
郝燕捷1,2,陈 莹1,薛 林1,韩晓宁1,丁文惠1*
(北京大学第一医院1.心内科;2.风湿免疫科,北京 100034)
目的 研究白细胞介素10(IL-10)对转化生长因子β1(TGF-β1)诱导的大鼠心脏成纤维细胞(CFBs)增殖、向肌样成纤维细胞(MyoFbs)转化的影响及其作用通路。方法 原代培养SD乳鼠CFBs,应用第2~4代,分为对照组、IL-10刺激组、TGF-β1刺激组、IL-10与TGF-β1共同刺激组(IL-10预处理后加入TGF-β1)。每个刺激组设3个复孔,所有实验重复3次。四氮唑盐比色法检测细胞增殖,免疫细胞化学染色检测增殖细胞核抗原表达,免疫细胞化学染色及Western blot检测α-平滑肌细胞肌动蛋白(α-SMA)和ERK1/2、P38激酶磷酸化蛋白的表达。结果 与对照组比较,TGF-β1(10 μg/L)能显著刺激 CFBs增殖及表型转化(α-SMA 表达)(P<0.01),用 IL-10(10、50和100 μg/L)预处理后,TGF-β1诱导的CFBs增殖及α-SMA表达呈剂量依赖性被显著抑制(P<0.01)。TGF-β1能显著刺激CFBs内ERK1/2和P38磷酸化水平(P<0.01),而IL-10(100 μg/L)预处理后,TGF-β1诱导的ERK1/2和P38激酶磷酸化被明显抑制(ERK1/2:P<0.05;P38:P<0.01)。结论 IL-10可能通过抑制ERK1/2和P38激酶活化抑制了TGF-β1诱导的CFBs增殖及向MyoFbs转化。
白细胞介素-10;心脏成纤维细胞;转化生长因子-β1;细胞外信号调节激酶;P38激酶
在急性心肌梗死(acute myocardial infarction,AMI)后的心室重构发展过程中,心脏成纤维细胞(cardiac fibroblasts,CFBs)增殖、向肌样成纤维细胞(myofibroblasts,MyoFbs)转化、分泌大量细胞外基质是重要的病理机制之一。转化生长因子-β1(transforming growth factor-β,TGF-β1)能够促进 CFBs 增殖及表型转化,是AMI后左室重构重要促进因子[1]。
白介素10(interleukin-10,IL-10)是一种多功能性免疫调节细胞因子,在抑制纤维化方面也有重要作用,它能拮抗 TGF-β1 促进肝脏纤维化[2]。在AMI患者的血浆及心肌组织中IL-10有高表达,而且能够改善AMI大鼠的左室重构[3]。IL-10能否抑制CFBs的增殖/表型转化及拮抗TGF-β1的作用,目前尚未见报道。
丝裂原活化蛋白激酶(MAPKs)超家族成员ERK1/2及P38在TGF-β1诱导的CFBs表型转化中发挥了重要作用[4],IL-10能够抑制 MAPKs通路[5]。本研究观察 IL-10 对 TGF-β1 诱导的大鼠CFBs增殖及向MyoFbs表型转化的影响,及是否通过ERK1/2及P38通路调控。
1 材料与方法
1.1 材料
实验动物选用清洁级SD乳鼠[北京大学第一医院实验动物中心提供,合格证号:SYXK(京)2014-0010];重组大鼠 IL-10及重组人 TGF-β1(Peprotech公司);小鼠抗人α-平滑肌肌动蛋白(α-SMA)单克隆抗体(武汉博士德公司);小鼠抗人增殖细胞核抗原(PCNA)单克隆抗体及免疫细胞化学染色试剂盒PV-6001/6002 PowerVisionTM(北京中杉金桥生物技术有限公司);兔抗大鼠ERK1/2、磷酸化ERK1/2、P38和磷酸化 P38多克隆抗体(Cell Signaling公司);ERK1/2及 P38的抑制剂 PD98059和SB203580(Promega公司)。
1.2 方法
1.2.1 原代培养SD乳鼠CFBs及鉴定:将乳鼠心室剪碎,0.1%胰蛋白酶消化、离心、收集细胞于DMEM培养液中,差速贴壁法培养30 min,贴壁的主要是心脏成纤维细胞。继续培养至细胞接近汇合时以1∶3传代。实验采用第2~4代。
免疫细胞化学染色对细胞进行鉴定:Vimentin染色阳性而α-SMA染色阴性证实为成纤维细胞。
1.2.2 IL-10对TGF-β1诱导的CFBs增殖及表型转化的影响:实验分组:1)对照组(control):培养过程中不加任何刺激剂;2)IL-10刺激组(10、50和100 μg/L3 个亚组);3)TGF-β1 刺激组:10 μg/L;4)IL-10(10、50 和 100 μg/L)预处理 1 h 后再加TGF-β1(10 μg/L)共同刺激组。48 h 后进行下面实验。
MTT比色法:参照文献[6]进行。
免疫细胞化学染色法测定PCNA及α-SMA表达:按试剂盒说明书操作。在MTT实验结果基础上IL-10选取100 μg/L剂量。染色之后,每张爬片在400倍倒置相差显微镜下拍摄4个无重复视野,用Image-Pro Plus 5.0图像分析系统计数细胞核呈棕色的PCNA阳性细胞核及所有细胞核,计算增殖指数:增殖指数(proliferation index)(%)=(PCNA阳性染色细胞核数/所有细胞核数)×100%。计数胞质染成黄色的α-SMA阳性细胞数及所有细胞数,计算转化指数:转化指数(transformation index)=(α-SMA阳性染色细胞数/所有细胞数)×100%。
1.2.3 TGF-β1对 ERK1/2、P38 磷酸化的影响:实验分组:1)对照组(control):即0 min组;2)TGF-β1刺激组:分别刺激 30 min及 1、2、4、8和 24 h。Western blot测定ERK1/2及 P38总蛋白及磷酸化表达,根据文献[5]进行。
1.2.4 应用ERK1/2、P38激酶阻断剂后TGF-β1对成纤维细胞表型转化的影响:实验分组:1)对照组(control);2)TGF-β1刺激组;3)PD98059(ERK1/2阻断剂)组:50 μmol/L;4)SB203580(P38阻断剂)组:20 μmol/L;5)TGF-β1+PD98059 组:先 用PD98059(50 μmol/L)预处理 1 h 后再与 TGF-β1 共同刺激;6)TGF-β1+SB203580组:先用预处理SB203580(20 μmol/L)1 h 后再与 TGF-β1 共同刺激。刺激24 h后收集细胞蛋白进行实验检测。Western blot测定 α-SMA蛋白表达,结果以相对α-SMA表达(α-SMA/β-actin吸光度比值)表示。
1.2.5 IL-10 对 TGF-β1 活化的 ERK1/2、P38 激酶的影响:实验分组:1)对照组(control);2)IL-10刺激组:100 μg/L;3)TGF-β1 刺激组;4)IL-10(100 μg/L)预处理1 h后再TGF-β1共同刺激组。Western blot测定ERK1/2及P38蛋白表达。ERK1/2组于TGF-β1刺激4 h后收集蛋白;P38组于进行TGF-β1刺激2 h后收集蛋白进行检测。结果以相对ERK1/2或P38表达(ERK1/2或P38/β-actin吸光度比值)表示。
上述实验n=3,重复3次。
1.3 统计学分析
采用SPSS 11.5软件进行统计分析。计量资料以均数±标准差(±s)表示,组间比较采用单因素方差分析,两两比较采用LSD检验。
2 结果
2.1 IL-10抑制TGF-β1诱导的CFBs增殖
2.1.1 MTT结果:TGF-β1组 A值显著增加(P<0.01),用IL-10与TGF-β1共同刺激后,细胞增殖较单纯 TGF-β1 刺激组下降(10 μg/L 及 50 μg/L 组P<0.05;100 μg/L组P<0.01),抑制作用随IL-10浓度增加而有所增强(图1)。在不加用TGF-β1而单独用IL-10刺激的状态下,各个IL-10剂量组细胞增殖较对照组均无明显变化(各组间比较P>0.05,图2)。
2.1.2 PCNA染色结果:TGF-β1组细胞增殖指数显著增加(P< 0.01),IL-10(100μg/L)+TGF-β1组增殖指数显著低于TGF-β1组(P<0.01)(图3)。
2.2 L-10浓度依赖性地抑制TGF-β1诱导的CFBs表型转化
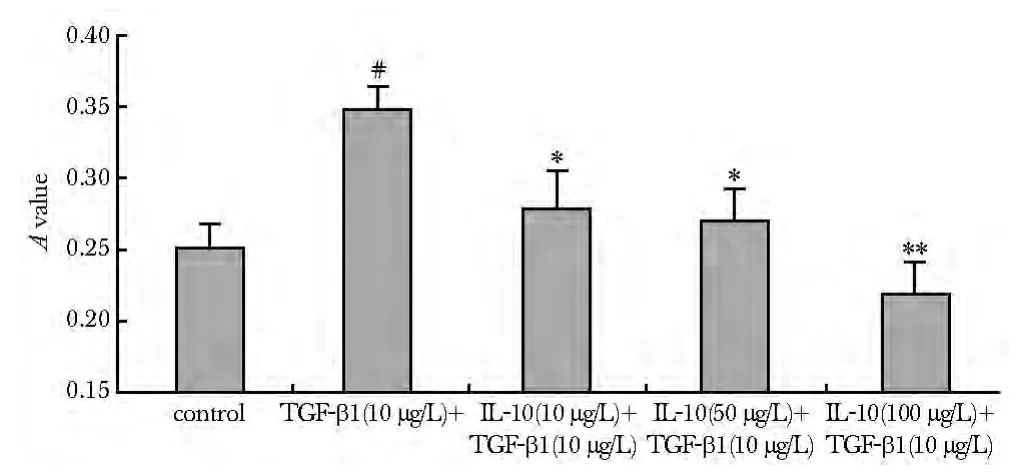
图1 IL-10对TGF-β1诱导的心脏成纤维细胞增殖的影响Fig 1 Effect of IL-10 on cardiac fibroblasts proliferationinduced by TGF-β1(±s,n=9/group)
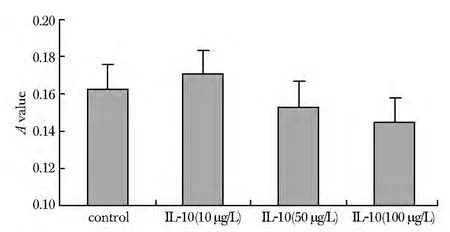
图2 IL-10对基础状态下心脏成纤维细胞增殖的影响Fig 2 Effect of IL-10 on cardiac fibroblasts proliferationunder basal condition(±s,n=9/group)
TGF-β1刺激组较对照组α-SMA表达阳性细胞比率显著增多(P<0.01);但IL-10+TGF-β1组较单纯TGF-β1组,α-SMA阳性表达明显减少(10 μg/L组P<0.05;50 μg/L 及 100 μg/L 组:P<0.01),且抑制作用随IL-10浓度增加而增强(图4)。
2.3 TGF-β1及 IL-10对 ERK1/2和 P38磷酸化的影响
2.3.1 TGF-β1诱导 ERK1/2及 P38磷酸化:ERK1/2在加入TGF-β1刺激后30 min时即发生明显磷酸化,4 h磷酸化水平最高,维持至8 h,24 h恢复至基础水平;P38于TGF-β1刺激后2 h磷酸化程度最高,随后磷酸化水平逐渐降低,24 h恢复基础水平(图5)。
2.3.2 阻断ERK1/2、P38激酶活性TGF-β1诱导的α-SMA 表达降低:与 TGF-β1 组相比,50 μmol/L PD98059预处理1 h再与TGF-β1共孵育组α-SMA蛋白的表达显著降低(P<0.05),20 μmol/LSB203580预处理组α-SMA蛋白的表达也显著降低(P<0.01)(图6)。
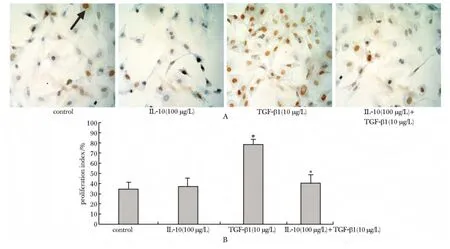
图3 IL-10对基础状态及TGF-β1诱导的心脏成纤维细胞增殖的影响Fig 3 Effects of IL-10 on the PCNA expression of cardiac fibroblasts under basal and TGF-β1-induced conditions
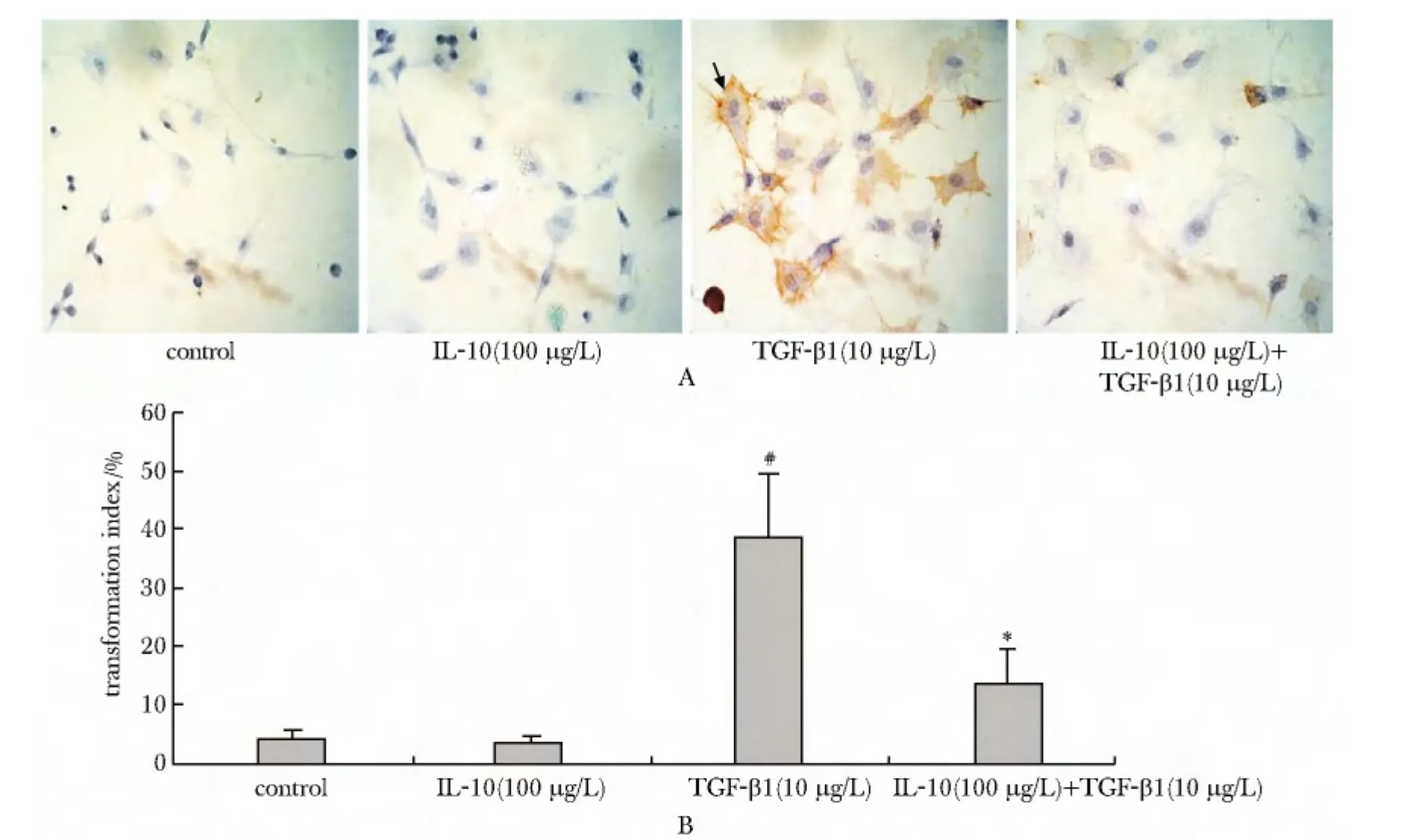
图4 IL-10对基础状态及TGF-β1诱导的心脏成纤维细胞向肌样成纤维细胞转化的影响Fig 4 Effects of IL-10 on the α-SMA expression of cardiac fibroblasts under basal and TGF-β1 induced conditions measured by immunocytochemical staining(×400)
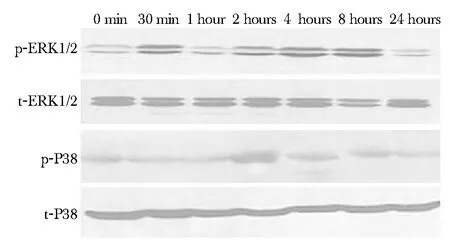
图5 ERK1/2、P38蛋白在TGF-β1刺激后各时间点磷酸化蛋白表达情况Fig 5 Effects of TGF-β1 on the phosphorylation of ERK1/2 and P38 in cardiac fibroblasts measured by Western blot
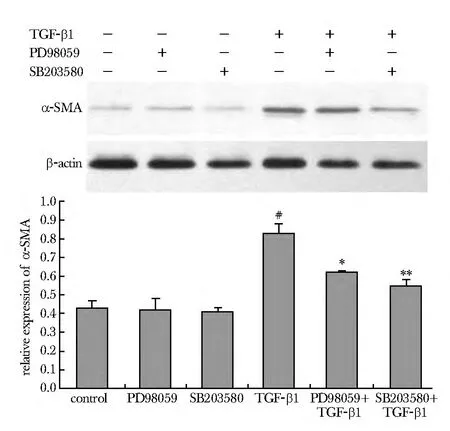
图6 ERK1/2及P38抑制剂对TGF-β1诱导的α-SMA蛋白表达的影响Fig 6 Effects of TGF-β1 on the α-SMA expression of cardiac fibroblasts when ERK1/2 and P38 pathway were inhibited
2.3.3 与TGF-β1组相比,IL-10抑制了 TGF-β1诱导的ERK1/2和P38激酶磷酸化 IL-10(100 μg/L)+TGF-β1组ERK1/2及P38磷酸化水平均显著低于 TGF-β1刺激组(ERK1/2:P<0.05;P38:P<0.01)(图7)。
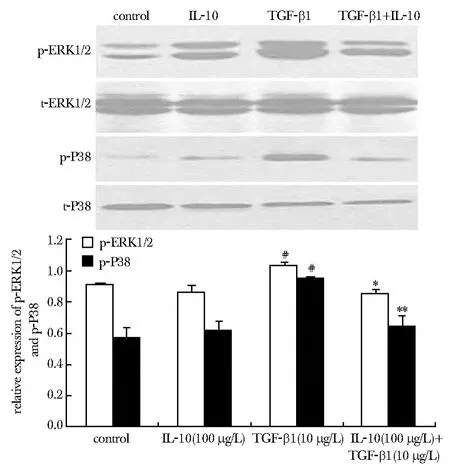
图7 IL-10对TGF-β1诱导的ERK1/2及P38磷酸化蛋白表达的影响Fig 7 Effects of IL-10 on the phosphorylation of ERK1/2 and P38 stimulated by TGF-β1 in cardiac fibroblasts
3 讨论
在大鼠 AMI模型上发现,在 AMI后 TGF-β1 mRNA在心肌中的表达显著增加,与其相伴随的是MyoFbs增多及胶原的堆积[7]。本结果显示TGF-β1刺激后CFBs增殖显著增强,同时向MyoFbs转化增多,进一步从细胞水平证实了TGF-β1在AMI后促纤维化的作用机制。
在缺血再灌注心肌组织中IL-10 mRNA及蛋白表达显著升高,IL-10能够改善心肌梗死大鼠的左室重构[3],能够抑制大鼠梗死后心肌细胞外Ⅰ、Ⅲ型胶原的沉积[8]。但具体机制尚不清楚。本研究证实IL-10对于基础状态下的CFBs无显著影响,但能够抑制TGF-β1促CFBs增殖的作用,且此作用呈浓度依赖趋势。
既往在IL-10与成纤维细胞表型转化方面仅见少数研究报道:IL-10能够抑制人类蜕膜间质细胞(一种类肌样成纤维细胞)α-SMA 的表达[9];IL-10-/-小鼠损伤愈合明显较正常对照组快,且表达α-SMA的MyoFbs数量明显增多[10]。本实验结果在细胞水平证实了IL-10能够抑制TGF-β1所诱导的CFBs向MyoFbs的转化。
本实验发现在体外IL-10能够抑制TGF-β1诱导的CFBs及向MyoFbs表型转化,这可能是AMI后IL-10抑制心室重构的作用机制之一。
研究显示AMI后ERK1/2和P38通路的活化与心室重构关系密切,而一系列研究证实几种MAPKs(ERK1/2及P38等)都能够被TGF-β1迅速激活[11-12],本实验证实 TGF-β1 能够促进 ERK1/2及P38的磷酸化,而ERK1/2及P38的抑制剂能够显著减少TGF-β1所诱导的α-SMA表达,更进一步证实了TGF-β1与MAPKs之间的生物联系。IL-10的经典作用信号通路是JAK-STAT通路系统,但有研究发现IL-10改善心肌梗死大鼠左室重构的作用部分是通过抑制P38激酶发挥的[3],本实验室的前期研究显示IL-10能够通过抑制此通路抑制Ⅰ、Ⅲ型胶原的合成。本研究结果进一步从细胞水平证实了IL-10能够抑制 TGF-β1诱导的ERK1/2及 P38磷酸化,提示抑制ERK1/2及P38通路可能是IL-10抑制TGF-β1作用的可能机制。
本研究显示IL-10可能是通过抑制ERK1/2和P38激酶活化抑制TGF-β1诱导的CFBs的增殖及表型转化。
[1]van Nieuwenhoven FA,Turner NA.The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction [J]. Vascul Pharmacol,2013,58:182-188.
[2]Huang YH,Chen YX,Zhang LJ,et al.Hydrodynamicsbased transfection of rat interleukin-10 gene attenuates porcine serum-induced liver fibrosis in rats by inhibiting the activation of hepatic stellate cells [J].Int J Mol Med,2014,34:677-686.
[3]Krishnamurthy P,Rajasingh J,Lambers E,et al.IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR [J].Circ Res,2009,104:e9-18.DOI:10.1161/CIRCRESAHA.108.188243.
[4]Liu XY,Sun SQ,Hassid A,et al.cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts[J].Mol Pharmacol,2006,70:1992-2003.
[5]Dhingra S,Sharma AK,Singla DK,et al.P38 and ERK1/2 MAPKs mediate the interplay of TNF-alpha and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis[J].Am J Physiol Heart Circ Physiol,2007,293:H3524-3531.
[6]Mosmann T.Mosmann T.Rapid colorimetric assay for cellular growth and survival:application to proliferation and cytotoxicity assays[J].J Immunol Methods,1983,65:55-63.
[7]Sun Y,Zhang JQ,Zhang J,et al.Cardiac remodeling by fibrous tissue after infarction in rats[J].J Lab Clin Med,2000,135:316-323.
[8]韩晓宁,胡春阳,褚松筠,等.白细胞介素-10抑制大鼠急性心肌梗死后左室心肌胶原沉积[J].基础医学与临床,2010,30:6-12.
[9]Kimatrai M,Blanco O,Muñoz-Fernández R,et al.Contractile activity of human decidual stromal cells.II.Effect of Interleukin-10[J].J Clin Endocrinol Metab,2005,90:6126-6130.
[10] Eming SA,Werner S,Bugnon PH,et al.Accelerated wound closure in mice deficient for interleukin-10[J].Am J Pathology,2007,170:188-202.
[11]Mulder KM.Role of Ras and Mapks in TGF beta signaling[J].Cytokine Growth Factor Rev,2000,11:23-35.
[12]Massagué J,Blain SW,Lo RS.TGF-β signaling in growth control,cancer, and heritable disorders [J].Cell,2000,103:295-309.
IL-10 inhibits cardiac fibroblasts proliferation and phenotype transformation induced by TGF-β1 in rats
HAO Yan-jie1,2,CHEN Ying1,XUE Lin1,HAN Xiao-ning1,DING Wen-hui1*
(1.Dept.of Cardiology;2.Dept.of Rheumatology and Clinical Immunology,the First Hospital,Peking University,Beijing 100034,China)
ObjectiveTo examine the effects of IL-10 on cardiac fibroblasts(CFBs)proliferation and phenotype transformation to myofibroblasts(MyoFbs)induced by transforming growth factor-β1(TGF-β1);and to investigate the regulating pathways.MethodsCardiac fibroblasts were isolated from cardiac ventricles of neonatal SD rats.The passage 2~4 were used and divided into the following groups for treatment:1)control group,2)IL-10 reaction group,3)TGF-β1 reaction group,and 4)IL-10 plus TGF-β1 reaction group(TGF-β1 treatment followed with IL-10 pretreatment).Cells proliferation was assessed by MTT assay and immunocytochemistry staining for proliferating cell nuclear antigen(PCNA);the phenotype transformation into MyoFbs was assessed by immunocytochemistry of α-smooth muscle actin(α-SMA);extracellular signal related kinase(ERK1/2)and P38 kinase pathways were assessed by western-blot.ResultsTGF-β1(10 μg/L)treatment boosted the proliferation and the expression of α-SMA significantly(P<0.01),while IL-10(10,50 or 100 μg/L)plus TGF-β1 co-treatment induced lower cell proliferation and expression of α-SMA than treating with TGF-β1 alone(P< 0.05),with the inhibitory effect of IL-10 being concentration dependent.TGF-β1 could significantly stimulate the ERK1/2 and P38 kinase phosphorylation(P<0.01),however IL-10(100 μg/L)plus TGF-β1 co-treatment failed to down-regulated the phosphorylation of ERK1/2 and P38 kinase compared with TGF-β1 alone(ERK1/2:P<0.05;P38:P<0.01).ConclusionsIL-10 can attenuate TGF-β1-induced CFBs proliferation and phenotype transformation to MyoFbs.The inhibitory effects may explained by a mechanism of inhibiting the activation of ERK1/2 and P38 kinase.
interleukin-10;cardiac fibroblasts;transforming growth factor-β1;extracellular signal related kinase;P38 kinase
R541.1
A
10.16352/j.issn.1001-6325.2015.09.007
1001-6325(2015)09-1182-06
2015-01-09
2015-04-27
国家自然科学基金(30640083)
*通信作者(corresponding author):dwh_rd@126.com
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
天津医科大学学报(2021年3期)2021-07-21
云南医药(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
现代园艺(2017年21期)2018-01-03
中国医药生物技术(2015年4期)2015-12-26
中国康复理论与实践(2015年10期)2015-12-24
中国现代医学杂志(2015年26期)2015-12-23
医学研究杂志(2015年5期)2015-06-10
中国当代医药(2015年33期)2015-03-01
