
病原微生物感染的蛋白质组学研究
2015-06-01 10:55胡墨杨玉飞刘小云
大学化学 2015年3期
胡墨 杨玉飞 刘小云
(北京大学化学与分子工程学院分析化学研究所和合成与功能生物分子中心 北京 100871)
病原微生物感染的蛋白质组学研究
胡墨杨玉飞刘小云*通讯联系人,E-mail:xiaoyun.liu@pku.edu.cn
(北京大学化学与分子工程学院分析化学研究所和合成与功能生物分子中心北京 100871)
摘要基于质谱的蛋白质组学在近20年有巨大的发展。在其应用中,病原微生物和与感染相关的蛋白质组学具有重要科学意义,体系复杂度又相对较小,一直受到广泛关注并有较快发展。本文从感染中病原微生物和宿主的蛋白质组学两方面入手,简要综述应用蛋白质组学研究感染过程的相关工作,着重介绍该领域近几年的主要进展,并对其发展做出展望。
关键词蛋白质组学质谱病原微生物宿主感染
A Proteomic View of Host-pathogen Interactions
and Infection Biology
Hu MoYang YufeiLiu Xiaoyun*
(CollegeofChemistryandMolecularEngineering,InstituteofAnalyticalChemistryandSyntheticandFunctional
BiomoleculesCenter,PekingUniversity,Beijing100871,China)
AbstractMS-based proteomics has undergone rapid advancements in recent years. As infectious diseases cause millions of death each year, it has become increasingly important to study host-pathogen interactions on the molecular level. Recently, proteomic analyses of pathogens and their mammalian host have received widespread attentions. Here we review the major progress made in this area within the last decade.
Key WordsProteomics; Mass spectrometry; Pathogen; Host; Infection
1背景介绍
蛋白质组学最早的工作来自于对细菌蛋白组的研究[1]。1975年,Patrick O′Farrell使用二维聚丙烯酰胺凝胶电泳(2D-PAGE)对大肠杆菌(Escherichiacoli)蛋白进行分离,得到超过1000个凝胶点,标志着人类第一次实现了对生物体全部蛋白进行系统层次的分离[2]。随着基因组学的发展和人类基因组计划的进行,蛋白质组(proteome)这一概念最早于1996年被提出[3],指一个细胞或生物体内全部的蛋白质;分析蛋白质组的学科则被称为蛋白质组学(proteomics)[4]。蛋白质是生命活动的执行者,一切基因和转录层次的变化,最终都要在蛋白质的层次(如表达量和翻译后修饰)发生变化才能表现出其生命意义。因此,蛋白质组学一直受到生命科学研究者的广泛关注。
在基质辅助激光解吸(matrix assisted laser desorption ionization,MALDI)和电喷雾(electrospray ionization,ESI)这两种可电离多肽和蛋白质的离子化技术产生后,基于质谱的蛋白质组学(mass spectrometry-based proteomics)得到了快速发展[5-6]。多肽和蛋白质离子在一定的激发条件如碰撞诱导解离(collision induced dissociation,CID)或电子俘获解离(electron capture dissociation,ECD)等作用下,产生在肽链骨架上的断裂,即可通过多肽/蛋白质的多级质谱得到其结构信息。与传统分析化学和生物化学鉴定蛋白采用的氮端测序和免疫印迹等方法相比,基于质谱的蛋白质组学拥有分析速度快、样品用量小、序列覆盖率高、定量手段成熟、易于分析翻译后修饰等众多优点。因此,基于质谱的蛋白质组学成为当前蛋白质组学领域最主要也是发展最快的方法。
与高等真核生物的蛋白组相比,原核生物编码的蛋白质数量少得多,蛋白的复杂性(如RNA剪切造成的一个基因对应多个蛋白)也远远小于高等真核生物。从分析化学的角度来说,体系的复杂度相对较小,因此以原核生物及其他微生物作为模型体系的蛋白质组学相对发展较快。在研究微生物的蛋白质组学时,一个具有重要科学意义的问题是病原微生物和与感染(infection)相关的蛋白质组学。世界卫生组织的统计数据表明,病原微生物引发的传染病造成的死亡占2011年全部死亡原因的1/3,因此对病原微生物感染过程的研究也具有极其重要和迫切的现实意义。在病原微生物入侵和感染宿主以及宿主拮抗病原微生物感染的过程中,执行关键生命活动的蛋白质究竟有哪些?它们是如何发挥生命作用的?这些问题都只有在蛋白质层次进行研究才能得到最终的解答。通过对这些重要生命过程分子机制的理解,人们将对病原微生物-宿主相互作用的过程有更加深入的认识,从而为设计抗感染的药物以及抑制病原微生物引发的传染病提供思路。
学术期刊Proteomics在2011年已出版了关于微生物蛋白质组学的专刊[7],其中大部分工作与病原微生物有关。本文将从病原微生物和宿主两方面入手,简要介绍应用蛋白质组学研究病原微生物感染过程的相关工作,着重介绍该领域最近几年的主要进展,并对相关方面的发展做出展望。
2病原微生物的蛋白质组学
2.1 体外培养条件下病原微生物的蛋白质组学
在病原微生物-宿主相互作用体系中,病原微生物的蛋白组相对简单,因此病原微生物成为应用蛋白质组学研究这个体系时的首要目标。体外培养条件下获取大量病原微生物相对简单,早期相关研究主要集中在体外培养环境下的病原微生物的蛋白质组学。
Adkins等比较了野生型沙门氏菌(SalmonellaTyphimurium,LT2)和一种毒性更强的沙门氏菌亚型(14028)在对数生长期、稳定期和缺镁的基本培养基中的蛋白组[8],共鉴定到2343个蛋白。通过在系统层次比较两种不同基因型的沙门氏菌的蛋白表达组,他们发现一些受pdu启动子调控的蛋白在14028亚型中具有更高的表达量。他们的工作表明,pdu启动子对于沙门氏菌的致病性有着重要的意义。
Ansong等比较了野生型、敲除hfq或smpB基因的沙门氏菌在4种体外培养条件下蛋白质组和转录组的差异[9]。hfq和smpB两个突变株中20%和4%的蛋白表达发生了显著变化,生物信息学分析表明受调控的蛋白包括与入侵相关、细胞基础代谢、脂多糖(lipopolysaccharides,LPS)合成、脂肪酸合成等功能。
Becher等以稳定同位素(氮-15)代谢标记定量的方法鉴定了处于对数生长期和稳定期的金黄色葡萄球菌(Staphylococcusaureus)蛋白表达组[10]。为了提高蛋白鉴定的覆盖率,他们通过亚细胞分离的方法把金黄色葡萄球菌在体外条件下分成4个组分:胞浆蛋白、膜蛋白、细胞表面蛋白和胞外蛋白。最终他们鉴定到1703个蛋白(定量1450个蛋白),达到了对全蛋白组65%的蛋白覆盖率。
总结已有的工作,一些重要的病原微生物如沙门氏菌[8-9,11-13]、志贺氏菌[14-16](Shigellaflexneri)、绿脓杆菌[17-19](Pseudomonasaeruginosa)、幽门螺杆菌[20-25](Helicobacterpylori)、结核分枝杆菌[26-27](Mycobacteriumtuberculosis)、金黄色葡萄球菌[10]、炭疽芽孢杆菌[28](Bacillusanthracis)在体外培养条件下的蛋白质组学都已有相关报道。与植物病原真菌相关的蛋白质组学工作也已有详尽综述发表[29],本文不再赘述。
感染宿主前后,病原微生物的生存环境有显著的变化,因此病原微生物的应激反应是研究的一大重点。在体外培养中,病原微生物的代谢模式、生长速率控制以及刺激效应蛋白分泌(即模拟入侵宿主时的条件)下的蛋白调控一直是研究者关注的热点。常见的控制条件为不同培养条件(无氧、高盐、不同碳源、培养基中某些离子缺陷、温度)、不同生长阶段(对数期和平台期)等。其中通过模拟感染后体内环境的体外培养条件研究感染时病原微生物发生的重要变化(如代谢调整(碳源和某些离子的代谢)、群体感应系统[30](quorum sensing)等)通常是进一步生物研究的主要方向。通常的研究思路是通过蛋白质组学大规模表征病原微生物在不同培养条件下(或敲除某些关键基因后)的蛋白表达组变化,从而得出病原微生物的应激反应机理或其中关键蛋白的作用通路及信号网络。也有一些工作研究了病原微生物中某些修饰组的变化,详见Jones[31]和Macek[32]的综述。
2.2 感染过程中体内病原微生物的蛋白质组学
与大量宿主细胞相比,病原微生物的蛋白量通常远小于宿主;同时,入侵宿主的病原微生物的量通常远远小于体外培养。因此,在体内蛋白组的实验中,为了达到更高的选择性和灵敏度,首先要解决的问题是从宿主细胞中分离出体内病原微生物。目前主要有3种分离方法[33],即离心(centrifugation)、基于免疫磁珠分离(immunomagnetic separation,IMS)及流式细胞术(fluorescence-activated cell sorting,FACS)。
感染过程中体内微生物的蛋白质组学始于2006年Becker等的工作[34]。他们采用流式细胞术从受到沙门氏菌感染的小鼠体内分离出沙门氏菌。在他们的分离条件下,对沙门氏菌蛋白的鉴定受到宿主蛋白的极大干扰,在鉴定到的蛋白中,超过70%都是宿主的蛋白。但液质联用平台极强的分离鉴定能力仍使他们共鉴定到约700个沙门氏菌的蛋白。进一步的生物信息学分析表明,沙门氏菌的代谢中广泛存在着代谢冗余机制(metabolic redundancy),只有极少数酶是其代谢所必需的。因此抗感染的药物所能针对的目标代谢途径很有限。
关于体内病原微生物的蛋白质组学早期的发展,Schmidt等已在2011年进行了综述[33]。当前这一领域最大的不足之处是与体外培养相比,鉴定到的蛋白数量还相对较少,通常只有几百个蛋白,覆盖率仅10%~20%(图1)[35~37]。这对于从蛋白组数据中分析病原微生物入侵后采用的分子机制,发掘具有重要生物学意义的结果是远远不够的。
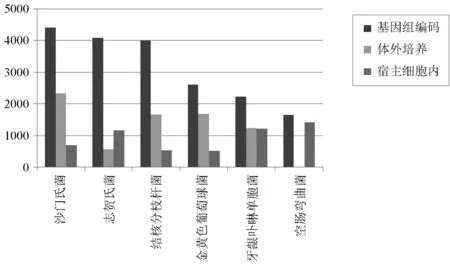
图1 几种病原微生物在体外培养和宿主细胞内鉴定到的蛋白数量与基因组理论编码蛋白数量的比较参考文献:沙门氏菌[8,34-35];志贺氏菌[14,38];结核分枝杆菌[26,36];金黄色葡萄球菌[10,37];牙龈卟啉单胞菌[39];空肠弯曲菌[40]
随着胞内病原微生物分离过程的优化及更高灵敏度的液质联用平台的应用,从数量极其有限的胞内微生物中进行高覆盖率蛋白组分析逐渐成为可能[38-40]。空肠弯曲菌(Campylobacterjejuni)是一种重要的造成食源性疾病的细菌。Liu等通过定量蛋白组分析感染COS-1细胞的空肠弯曲菌蛋白组。他们采用离心方法分离胞内细菌,取得了较好的分离效果(宿主细胞干扰小于15%)[40],鉴定到1428个蛋白,覆盖率达到86%。通过高覆盖率的体内蛋白组数据,他们发现空肠弯曲菌入侵宿主细胞后,代谢水平发生了显著下降,并且重新调整了呼吸模式,更倾向于采用富马酸作为电子给体。他们的结果有助于人们针对胞内病原微生物特殊的代谢途径发展应对策略。
Pieper等采用类似方法研究了另一种重要病原微生物——志贺氏菌在宿主细胞体内的蛋白组变化[38]。他们采用超速离心的方法分离体内细菌,对比了体外培养,胞内和与细胞共同培养但不具备入侵能力的志贺氏菌的蛋白组。定量蛋白组数据表明志贺氏菌在宿主细胞内所用的主要碳源发生了明显变化,并且与摄取铁离子相关的蛋白发生了明显上调,相关蛋白突变体的进一步验证表明感染后志贺氏菌在宿主体内的增殖依赖于其获取铁离子的量。
绝大多数对于病原微生物的研究都以在体内实验获得在感染过程中的相关机制作为最终的目标,关于病原微生物的蛋白组研究同样如此。相信随着体内病原微生物分离和蛋白质组学液质联用平台的进一步发展,将实现在体内鉴定到与体外培养条件下数量相近的蛋白。从而通过更具深度和广度的蛋白质组学数据,给出病原微生物感染宿主后启动的重要分子机制,并为特异性抑制感染提供新思路。
3感染过程中宿主的蛋白质组学
与病原微生物相对简单的蛋白组相比,宿主通常是高等真核生物,其基因数量和翻译出的蛋白组的复杂度均远远高于前者。对宿主细胞进行高通量高覆盖率的定量蛋白质组学分析在技术上是更大的挑战。
对宿主蛋白表达组的研究可按照分析的宿主蛋白目标不同,分为蛋白表达组(包括全蛋白组、亚细胞蛋白组)和修饰组两类。亦可根据实验条件的不同分为两类:以病原微生物的某些模型代谢产物(如革兰氏阴性菌的脂多糖)或病原菌组成部分(如鞭毛)刺激;以病原微生物(野生型或基因缺陷型)感染。
3.1 表达组
Dhungana等应用脂多糖刺激巨噬细胞,采用稳定同位素氨基酸代谢标记(stable isotope labeling by amino acids in cell culture, SILAC)进行蛋白定量[41]。泛素-蛋白酶体系统在脂多糖刺激后重新分布到脂筏区(lipid raft region);MEK-ERK通路的激活也对蛋白酶体激活有调控作用。
Du等采用SILAC定量研究了LPS刺激仅10分钟后的宿主细胞蛋白组[42]。通过亚细胞分离,有效分离出胞浆和细胞核两个组分,从而提高了蛋白组鉴定的深度和广度。胞浆和细胞核中各有611和758个蛋白在LPS刺激10分钟后表达量发生显著变化。通过通路分析,他们发现MAPK和NF-κB通路在刺激10分钟后即发生了显著变化,说明宿主细胞对于病原微生物的应激反应是十分迅速的。
中性粒细胞(neutrophil)是免疫系统的重要组成部分,而中性粒细胞网具有降解效应蛋白、消灭革兰氏阴性菌及革兰氏阳性菌的作用。Urban等提取并分析了中性粒细胞网的蛋白组成,发现了24个与中性粒细胞网相关的蛋白[43]。他们在其中发现了钙卫蛋白(calprotectin)。进一步的体外和体内实验表明,钙卫蛋白确实参与了中性粒细胞网的形成。
Hardwidge等分析了野生型和三型分泌系统(type Ⅲ secretion system)缺失的突变型致病性大肠杆菌(EnteropathogenicEscherichiacoli)感染后Caco-2细胞系的蛋白组[44]。在鉴定到的2090个宿主蛋白中,264个的表达量有显著差异。
Backert等则结合转录组学与蛋白质组学的手段分析野生型及缺失四型分泌系统的幽门螺旋杆菌感染后的人胃上皮细胞[45]。他们采用二维凝胶电泳分离,鉴定到190个发生显著变化的蛋白。其中161个蛋白在感染后mRNA水平发生显著变化。他们的工作验证了四型分泌系统在幽门螺旋杆菌感染过程中的重要作用。
Meissner等人分析了脂多糖刺激后小鼠巨噬细胞的分泌蛋白组[46]。他们的研究包括5个不同的时间点和4种基因型的小鼠(WT/MyD88-ko/Trif-ko/MyD88-ko &Trif-ko),并同时分析了分泌组和巨噬细胞转录组的变化。在受到LPS刺激后,免疫细胞分泌蛋白组发生了极其显著的变化,部分蛋白分泌量的变化超过了1000倍。而敲除两个TLR4重要的接头蛋白MyD88或Trif后,巨噬细胞对于刺激的响应明显减弱。除了证明两个接头蛋白对于TLR4的信号传递通路引起的下游分泌蛋白的变化具有重要作用外,作者还通过对比单双敲除体的实验结果证实,两个接头蛋白对下游分泌蛋白的调控具有冗余作用、协同作用等几种不同的方式。
3.2 修饰组
除了表达量不同,不同的翻译后修饰也会极大地影响蛋白质的功能[47]。Sharma等研究了脂多糖刺激不同时间后巨噬细胞的磷酸化蛋白组[48]。大约140个磷酸化位点(175个磷酸化肽)在LPS刺激后发生了显著变化。大规模磷酸化蛋白组的实验结果对于发现TLR4通路相关蛋白有着重要作用。另外,通过生物信息学分析结合时间分辨的磷酸化蛋白组数据,人们对于LPS刺激后的巨噬细胞的不同时间、空间上不同细胞器相关蛋白的响应有了新的认识。
Rogers等研究了沙门氏菌感染后宿主细胞磷酸化蛋白组的变化[49]。通过未感染/野生型感染/缺失效应蛋白SopB的沙门氏菌的对照,他们从系统层面研究了效应蛋白SopB对宿主磷酸化蛋白组的影响,实验表明感染后宿主细胞磷酸化水平的变化之中有大约一半与SopB有关,说明SopB对于宿主细胞信号转导有着广泛的调控。
在对感染过程中宿主细胞蛋白组的研究方面,以下几个问题受到广泛关注[50]:宿主细胞对病原微生物的感受机制及受体蛋白;病原微生物效应蛋白在宿主细胞内的作用目标;病原微生物引起的宿主细胞信号传递链的变化;宿主细胞的免疫相关反应(如抗体的产生、免疫相关信号通路和分泌蛋白的变化)等。
4总结和展望
随着病原微生物蛋白质组学技术的发展,早期围绕着体外培养病原微生物和蛋白鉴定的工作已经不再成为主流,而研究感染后体内病原微生物和宿主细胞的工作在近几年不断增加。通过在系统层次高通量高覆盖率分析感染后病原微生物和宿主蛋白组的变化,我们将更加深入地认识在感染过程之中二者分别使用的主要分子机制。另外,结合多种蛋白质组学手段(如修饰组、分泌组、蛋白相互作用组)及上下游组学手段(转录组、代谢组)并互相对照[9,28,46],也会极大地推动人们对于病原微生物感染过程的认识。最终,以后续生物学实验进一步验证前期组学实验得到的初步结论及其功能,也将进一步促成蛋白质组学从产生组学数据向解答重要生命科学问题的重要转变。
参考文献
[1] Neidhardt F C.Proteomics,2011,11(15):2943
[2] O′Farrell P H.JBiolChem,1975,250(10):4007
[3] Wilkins M R,Pasquali C,Appel R D,etal.NatBiotech,1996,14(1):61
[4] James P.QuarterlyReviewsofBiophysics,1997,30(4):279
[5] Aebersold R,Mann M.Nature,2003,422(6928):198
[6] Altelaar A F M,Munoz J,Heck A J R.NatRevGenet,2013,14(1):35
[7]Proteomics,2011,11(15):2941
[8] Adkins J N,Mottaz H M,Norbeck A D,etal.MolCellProteomics,2006,5(8):1450
[9] Ansong C,Yoon H,Porwollik S,etal.PlosOne,2009,4(3):e4809
[10]Becher D,Hempel K,Sievers S,etal.PlosOne,2009,4(12):e8176
[11]Sonck K A J,Kint G,Schoofs G,etal.Proteomics,2009,9(3):565
[12]Qi S Y,Moir A,O′Connor C D.JBacteriol,1996,178(16):5032
[13]Coldham N G,Woodward M J.JProteomeRes,2004,3(3):595
[14]Zhu L,Zhao G,Stein R,etal.MolCellProteomics,2010,9(6):1209
[15]Wei C,Yang J,Zhu J,etal.JProteomeRes,2006,5(8):1860
[16]Liao X,Ying T,Wang H,etal.Electrophoresis,2003,24(16):2864
[17]Nouwens A S,Beatson S A,Whitchurch C B,etal.Microbiology,2003,149(5):1311
[18]Nigaud Y,Cosette P,Collet A,etal.BiochimicaetBiophysicaActa(BBA)-ProteinsandProteomics,2010,1804(4):957
[19]Hare N J,Scott N E,Shin E H H,etal.Proteomics,2011,11(15):3056
[20]Jungblut P R,Bumann D,Haas G,etal.MolMicrobiol,2000,36(3):710
[21]Lock A S J C,Coombs G W,Walsh B J,etal.Pathology,2001,33(3):365
[22]Bumann D,Aksu S,Wendland M,etal.InfectImmun,2002,70(7):3396
[23]Cho M-J,Jeon B-S,Park J-W,etal.Electrophoresis,2002,23(7-8):1161
[24]Kim N,Weeks D L,Shin J M,etal.JBacteriol,2002,184(22):6155
[25]Uwins C,Deitrich C,Argo E,etal.Electrophoresis,2006,27(5-6):1136
[26]de Souza G A,Fortuin S,Aguilar D,etal.MolCellProteomics,2010,9(11):2414
[27]Mehaffy M C,Kruh-Garcia N A,Dobos K M.JProteomeRes,2011,11(1):17
[28]Pohl S,Tu W Y,Aldridge P D,etal.Proteomics,2011,11(15):3036
[29]Gonzalez-Fernandez R,Jorrin-Novo J V.JProteomeRes,2011,11(1):3
[30]Eberl L,Riedel K.Proteomics,2011,11(15):3070
[31]Jones J D,O′Connor C D.Proteomics,2011,11(15):3012
[32]Macek B,Mijakovic I.Proteomics,2011,11(15):3002
[33]Schmidt F,Völker U.Proteomics,2011,11(15):3203
[34]Becker D,Selbach M,Rollenhagen C,etal.Nature,2006,440(7082):303
[35]Shi L,Adkins J N,Coleman J R,etal.JBiolChem,2006,281(39):29131
[36]Kruh N A,Troudt J,Izzo A,etal.PlosOne,2010,5(11):e13938
[37]Schmidt F,Scharf S S,Hildebrandt P,etal.Proteomics,2010,10(15):2801
[38]Pieper R,Fisher C R,Suh M-J,etal.InfectImmun,2013,81(12):4635
[39]Xia Q,Wang T,Taub F,etal.Proteomics,2007,7(23):4323
[40]Liu X,Gao B,Novik V,etal.PLoSPathog,2012,8(3):e1002562
[41]Dhungana S,Merrick B A,Tomer K B,etal.MolCellProteomics,2009,8(1):201
[42]Du R,Long J,Yao J,etal.JProteomeRes,2010,9(4):1805
[43]Urban C F,Ermert D,Schmid M,etal.PLoSPathog,2009,5(10):e1000639
[44]Hardwidge P R,Rodriguez-Escudero I,Goode D,etal.JBiolChem,2004,279(19):20127
[45]Backert S,Gressmann H,Kwok T,etal.Proteomics,2005,5(15):3902
[46]Meissner F,Scheltema R A,Mollenkopf H-J,etal.Science,2013,340(6131):475
[47]Choudhary C,Mann M.NatRevMolCellBiol,2010,11(6):427
[48]Sharma K,Kumar C,Kéri G,etal.JProteomeRes,2010,9(5):2539
[49]Rogers L D,Brown N F,Fang Y,etal.SciSignal,2011,4(191):rs9
[50]Hartlova A,Krocova Z,Cerveny L,etal.Proteomics,2011,11(15):3212
中图分类号O6;G64
doi:10.3866/pku.DXHX20150303
猜你喜欢
国外畜牧学(猪与禽)(2020年12期)2021-01-18
食管疾病(2019年2期)2019-07-03
国际口腔医学杂志(2019年3期)2019-05-31
食品与机械(2019年1期)2019-03-30
天然产物研究与开发(2018年2期)2018-04-04
广东蚕业(2016年9期)2016-01-17
医学研究杂志(2015年11期)2015-06-10
特产研究(2014年4期)2014-04-10
食品工业科技(2014年15期)2014-03-11
云南畜牧兽医(2014年4期)2014-02-28
