
利用Gateway技术改造原核表达载体pGEX—4T—1、pET—28a
2015-05-30 14:20胡丽松邬华松郝朝运谭乐和范睿吴刚
热带作物学报 2015年8期
关键词:原核表达
胡丽松 邬华松 郝朝运 谭乐和 范睿 吴刚

摘 要 质粒载体是基因工程研究中不可或缺的工具。为提高载体构建效率,进一步满足基因工程研究在构建表达载体时的需要,本文提供了一种重组型载体的改造策略。基本流程如下:利用带BglⅡ、XhoⅠ酶切位点以及attB重组序列的引物,从Gateway的入门载体pDONR Zeo中克隆到“BglⅡ-attB1-ccdB-attB2-XhoⅠ”片段。通过酶切连接的方法,将该片段插入到原核表达载体pGEX-4T-1以及pET-28a的多克隆位点,构建具有重组位点以及ccdB致死基因的原核表达载体。以胡椒High mobility group protein(HMGB)基因的开放读码框为目标片段,通过重组反应将目的基因构建到改造后的原核表达载体上,经检测体外诱导表达出大小正确的HMGB蛋白,验证本研究中改造载体的正确性。该载体的成功构建,为基因工程科研工作提供了一个高效的蛋白表达载体以及载体改造的新策略。
关键词 Gateway技术;载体改造;胡椒PnHMGB;原核表达
中图分类号 Q936 文献标识码 A
Modification of Prokaryotic Expression Vector
pGEX-4T-1, pET-28a by Using
Gateway Technology
HU Lisong1,2, WU Huasong1,2 *, HAO Chaoyun1,2, TAN Lehe1,2, FAN Rui1,2, WU Gang1,2
1 Spice and Beverage Research Institute,CATAS, Wanning, Hainan 571533, China
2 Ministry of Agriculture Key Laboratory of Genetic Resources Utilization of Spice and Beverage Crops / Hainan Provincial
Key Laboratory of Genetic Improvement and Quality Regulatioin for Tropical Spice and Beverage Crops,
Wanning, Hainan 571533, China
Abstract Plasmid vector is a necessary tools for gene engineering. In order to improve the efficiency of vector construction, a strategy for recombinant vector modification was presented. The process as follows: A“BglⅡ-attB1-ccdB-attB2-XhoⅠ”sequence was cloned with 5`-BglⅡ-attB1 and 3`-XhoⅠ-attB2 extension primers from Gateway entry vector pDONR Zeo. Then the sequence was inserted into the multiple cloning site of pGEX-4T-1 and pET-28a to construct the new recombinant ccdB-selection E. coli expression vector. The open read frame(ORF)sequence of PnHMGB was cloned to the new pGEX-4T-1 and pET-28a vector by recombinant reaction. The results of PnHMGB expression in vitro verifies the correctness of vector modification. The successful vector modification supply an efficient E. coli expression vector and a new strategy for recombinant vector modification.
Key words Gateway technology; Vector modification; PnHMGB; Prokaryotic expression
doi 10.3969/j.issn.1000-2561.2015.08.014
天然质粒是存在于受体细胞染色体外的一种裸露的小型环状双链DNA分子,广泛存在于细菌、霉菌、蓝藻、酵母等细胞,是寄主染色体以外的非必要组成部分,随着细胞的自我复制而分配到后代中。质粒还具有DNA转移性,能够使遗传物质从一细胞转移到另一细胞中。这些特点很好的符合了基因工程的研究需要,以质粒为骨架构建而成的载体统称为质粒载体[1]。目前构建的质粒载体有上百种,广泛应用的包括pCAMBIA系列真核表达载体,pET系列原核表达载体,pGEM-T克隆载体等[2]。
在质粒载体的基本元件中,有一段包含多个限制性酶切位点的DNA短序列,这一片段称为多克隆位点(multiple cloning site,MCS)或多位点接头(polylinker)。该元件是外源遗传物质导入载体的关键,通过合适的酶切位点,可以将一个或多个外源DNA片段插入到载体[3]。尽管目前多克隆位点标准配置序列的酶切位点足够丰富,但是实际的研究中会遇到目的基因片段与载体的酶切位点不匹配,致使目的基因片段插入载体相当困难[4]。另外,利用限制性内切酶和连接酶构建质粒载体时需要对DNA进行切割并回收目的片段,然后再用连接酶将其连接,这些操作使得传统的酶切连接克隆技术在应对大规模、高通量构建需求时具有一定的局限性[5]。
为了克服用酶切连接技术构建表达载体所遇到的问题,Invitrogen公司根据λ 噬菌体基因组和大肠杆菌基因组之间的位点专一性重组分子机制开发了一套分子克隆新技术,即Gateway克隆技术[6]。Gateway克隆技术是一种通用性的克隆方法,利用该技术可以快速、高效地将目的基因同时构建到多种与 Gateway 技术兼容的载体系统,用于功能分析和蛋白质表达[7-9]。它基于lambda噬菌体的位点特异性重组反应(attB×attP→attL×attR),在目的片段两端添加attB重组位点,将含attB重组位点PCR产物与含attP重组位点的入门载体混合,在BP酶的催化下发生反应,把目的基因克隆到入门载体上,同时在入门载体上形成新的attL位点。含新的attL位点的入门载体在LR重组酶的催化下和含attR位点的表达载体发生反应,从而达到将目的基因克隆到功能载体的目的[10-11]。与经典克隆多个步骤相比,该方法只需一步生化反应便能达到克隆目的,是高通量克隆基因的好方法。一旦构建获得包含目的基因的入门载体,这个入门克隆可以与任何Gateway下游目的载体进行LR重组反应,构建多种表达系统,用于基因超量表达、基因沉默、启动子分析、蛋白质亚细胞定位等研究[12-14]。另外,Gateway技术采用了致死基因ccdB的筛选方法,能确保高效率的分离阳性重组克隆[15]。尽管Gateway技术是高通量克隆基因的好方法,但由于专利等原因,Gateway后续表达系统不如经典的表达载体高效。
本研究以整合Gateway重组技术克隆的高效性和传统质粒表达的稳定性为目的,把带重组位点的序列插入到原核表达载体pET-28a和pGEX-4T-1上,构建具有重组位点及ccdB致死基因的原核蛋白表达载体。以胡椒HMGB开放读码框序列为目的片段,经两步重组构建到原核表达载体pET-28a和pGEX-4T-1上,通过体外IPTG诱导,分别获得带His和GST标签的蛋白,验证了该方法的可行性和正确性,为改造质粒载体提供一个新思路。
1 材料与方法
1.1 材料
质粒及菌株入门载体pDONR Zeo购自Life Technologies公司;原核表达载体pGEX-4T-1购自Amersham公司;pET-28a购自Novagen公司;菌株TOP10、DB3.1、Rosetta由华中农业大学作物遗传改良国家重点实验室棉花课题组惠赠。
主要生化试剂:限制性内切酶、T4连接酶、DNA扩增酶购自New England Biolabs(NEB)公司;BP、LR重组酶,Isopropyl β-D-1-Thiogalactopyranoside(IPTG)、抗生素购自Life Technologies公司;蛋白质电泳相关试剂购自伯乐(Bio-Rad)公司;PCR产物纯化、质粒提取试剂盒购自天根公司;其他试剂均为国产分析纯产品。
引物:实验中所设计的引物(表1)全部委托上海英骏生物技术有限公司(Invitrogen)合成。
1.2 方法
1.2.1 重组ccdB序列克隆 以BglⅡ-attB1序列为上游引物,XhoⅠ-attB2为下游引物,pDONR Zeo质粒为模板进行PCR扩增(95 ℃预变性5 min;94 ℃变性1 min,60 ℃退火30 s,72 ℃延伸1 min,30个循环;72 ℃延伸10 min),获得“BglⅡ-attB1-ccdB-attB2-XhoⅠ”片段,经电泳检测大小准确后回收;回收产物克隆到pGEM-T载体上,转化大肠杆菌TOP10,提取阳性克隆质粒,用BglⅡ、XhoⅠ在37 ℃ 、buffer 2体系进行双酶切反应2 h,回收目的片段待用。
1.2.2 原核表达载体改造 用BamHⅠ和XhoⅠ酶切pET-28a、pGEX-4T-1质粒,酶切体系为BamHⅠ、XhoⅠ各1 μL,质粒10 μg,buffer 2 μL,补双蒸水至20 μL,37 ℃反应2 h,电泳检测后挖胶回收。将酶切、回收后的载体及“BglⅡ-aatB1-ccdB-attB2-XhoⅠ”重组片段在T4连接酶的作用下16 ℃反应10 h。反应产物热激转化大肠杆菌DB3.1感受态,通过博来霉素(100 μg/mL)筛选阳性菌落,经PCR验证后,抽提质粒,保存菌株(检测引物见表1)。
1.2.3 原核表达载体验证 根据研究室已构建的胡椒叶片cDNA文库信息,选择PnHMGB基因作为验证基因,该基因编码一个约18 ku的蛋白质[16]。设计带attB接头引物进行扩增, PCR产物通过天根试剂盒回收。用BP重组酶将回收的PnHMGB基因构建到入门载体pDONR Zeo,转化大肠杆菌TOP10,挑取阳性克隆送公司测序,并抽提对应阳性克隆质粒。阳性质粒分别与改造后的pET-28a及pGEX-4T-1质粒在LR重组酶的作用下将PnHMGB基因构建到表达载体上,转化大肠杆菌,挑选阳性克隆,并扩繁,提取质粒。
构建好的pET-28a-PnHMGB、pGEX-4T-1- PnHMGB质粒转化大肠杆菌表达菌株Rosetta(DE3),挑取阳性菌落,接入1 mL含抗生素(100 μg/mL)的LB培养基,37 ℃摇床培养过夜。取100 μL培养物转接入10 mL含抗生素(100 μg/mL)的LB培养基中,37 ℃震荡培养,待OD600在0.5~0.7时,取1 mL培养物为对照,其余的加入IPTG至终浓度为1 mmol/L,16 ℃震荡培养8 h。取1 mL诱导培养物及未诱导的培养物,离心收集菌体,用100 μL SDS聚丙烯酰胺溶液悬浮,高温裂解后进行SDS聚丙烯酰胺凝胶电泳检测。
2 结果与分析
2.1 重组型ccdB序列克隆
利用PCR扩增获得重组ccdB序列,结果显示重组ccdB片段大约1.8 kb,扩增产物与预期大小符合。将目标片段克隆到T载体,将含有目的序列的T载体进行BglⅡ和XhoⅠ双酶切,酶切产物片段经电泳检测与克隆片段大小一致(图1)。结果表明重组型ccdB序列被准确克隆,可用于进一步实验。
2.2 载体改造与验证
用BamHⅠ和XhoⅠ双酶切pET-28a及pGEX-4T-1质粒。将含BglⅡ和XhoⅠ酶切位点的重组ccdB序列分别插入到pET-28a和pGEX-4T-1,构建2个含重组位点的原核表达载体。转化大肠杆菌DB3.1,用通用引物pGEX-F、pGEX-R、T7pro、T7ter进行PCR检测,如图1所示,改造成功的原核表达载体比改造前大2 kb左右,通用引物扩增产物包含了300 bp左右的载体序列,因此PCR产物比重组ccdB序列稍大。这些结果表明重组ccdB序列已经成功插入到原核表达载体上(图1)。
2.3 胡椒PnHMGB原核表达载体构建及验证
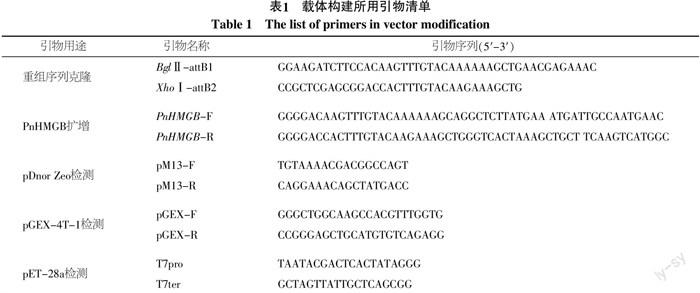
PCR扩增胡椒PnHMGB序列,测序结果显示,该基因开放读码框共432 bp,编码一个大小约为18 ku大小的蛋白(图2)。选择测序结果正确的克隆,扩增带重组位点的PnHMGB片段,在重组酶的作用下构建到pDONR Zeo上。抽提pDONR Zeo-PnHMGB质粒,在LR重组酶的作用下将胡椒PnHMGB序列从pDONR Zeo重组到pET-28a及pGEX-4T-1载体上。
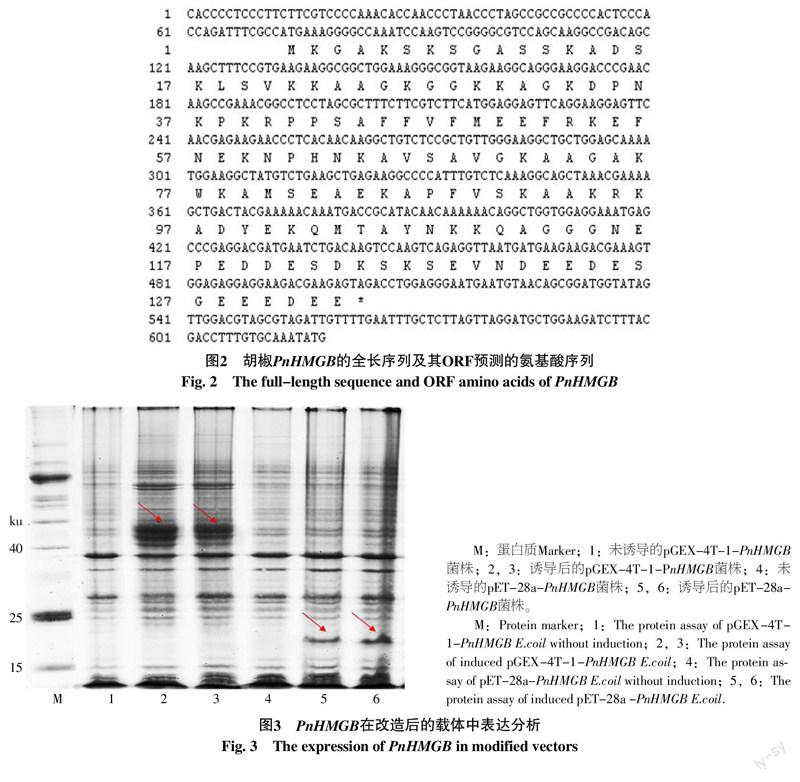
抽提重组正确阳性质粒,热激转化转化大肠杆菌表达菌株Rosetta(DE3),体外IPTG诱导PnHMGB表达。SDS-PAGE电泳结果表明:PnHMGB在pET-28a载体诱导出20 ku左右的特异表达蛋白,pGEX-4T-1载体诱导表达出40 ku左右的特异表达蛋白。正常的PnHMGB蛋白大小为18 ku,在pET-28a载体中表达末端带有6个His标签,所以大小应该为20 ku,而在pGEX-4T-1载体中表达末端带有23 ku大小的GST标签,因此大小应该为42 ku,如图3中红色箭头所示。通过2个载体的表达,确认PnHMGB能在改造后的载体中成功表达,表明本研究采取的重组型载体改造的方法可行。
3 讨论与结论
在基因功能验证的研究中,各种不同类型载体的构建是研究的基础之一。目前应用最为广泛的是基于酶切和连接反应的DNA 重组技术,如pCAMBIA真核表达系列,pET原核表达系列等。这类载体在表达效率和稳定性上得到研究者的肯定,目前被广泛应用在基因功能验证研究中。而在实际的应用中,研究者根据不同需求对这类载体进行一定的改造。潘求真等[17]利用双酶切将pEGFP-N1质粒上的多克隆位点(MSC)去除,然后补平连接, 获得增强型绿色荧光蛋白编码基因的真核表达载体pEGFP-N1-MSC。为获得无标记基因转基因植物、保证转基因生物安全,周红等[18]以pCAMBIA1300植物转化载体为骨架,通过AseI单酶切去除潮霉素标记基因,通过酶切片段的自连接反应构建了无标记基因的植物转基因载体。基于酶切连接的方法,在载体启动子改造、表达标签添加等方面被广泛应用[19-20],但是,这类载体的构建其中一个重要环节是选择合适的酶切位点。由于载体的多克隆位点是固定不变的,常常难以满足研究的具体需求,尽管稀有酶类,同尾酶的发现弥补了一些缺陷,整体构建的效率没有质的提升[5,21]。
基于位点特异性重组的Gateway 技术得到越来越多研究人员的青睐,该技术的应用使得外源基因的插入不再受酶切位点的限制,通过两步重组反应就能实现目的基因的插入,大大提高了载体构建效率[6-8,22]。本研究以整合Gateway重组载体的高效构建和传统载体的高效表达为目的,从研究的角度出发,展示了一种重组型原核表达载体的改造方法。该方法同样适用其他载体系统的改造,但是,在实际操作时需要注意以下几点:(1)Gateway系列载体核心重组和ccdB序列中包含了很多酶切位点,通过常规的方法很难将该序列转移到载体的多克隆位点。通过分析,我们选择了用BamHⅠ和XhoⅠ酶切载体,而用BglⅡ和XhoⅠ酶切重组序列,BamHⅠ和BglⅡ为同尾酶,酶切之后形成粘性末端可以在T4连接酶的作用下完成连接,从而达到转移目的。但是一旦连接成功,BamHⅠ和BglⅡ的位点就被破坏,无法通过再次酶切、转移[23];(2)连接后的载体因为含有ccdB致死基因,导致一般的DH5α、TOP10菌株无法扩繁,需选用特定DB3.1感受态进行扩繁[24]。(3)当载体完成改造后,必须通过目的基因进行表达验证,主要是因为致死基因ccdB的序列中含有高级结构,所以无法通过测序来确定读码框的正确,一旦在克隆过程中序列出现突变,则会影响到后续的表达研究。本研究选取胡椒PnHMGB作为目标基因验证载体的构建是否准确,是由该基因序列的特点而定。胡椒PnHMGB序列在蛋白翻译时存在3种读码可能,分别会翻译出18、9、6 ku等3种蛋白,但只有翻译出18 ku大小的蛋白才能说明载体按照正确的读码方式翻译蛋白,从而证明在改造过程中载体本身并没有发生突变、移码的情况。
通过重组序列克隆、转移到目的蛋白表达验证这一系列的实验操作,本文展示了一种重组型原核表达载体的改造方法,该载体的成功构建可以保证目标片段的插入不再受限于酶切位点的选择,同时引入ccdB致死基因的筛选方式大大提高了选择的效率。该研究为载体改造提供了新的思路,丰富并拓展了Gateway重组技术的应用。
参考文献
[1] 张献龙, 唐克轩. 植物生物技术[M]. 北京: 科学出版社, 2004.
[2] Sambrook J, Russell D W. 分子克隆实验指南[M]. 黄培堂, 黄恒樑, 周晓巍, 等译. 北京: 科学出版社, 2002.
[3] Dallas-Yang Q, Jiang G, Sladek F M. Digestion of terminal restriction endonuclease recognition sites on PCR products[J]. Biotechniques, 1998, 24(4): 582-584.
[4] 牛 麟. pTriEx-4neo载体多克隆位点的改造;抗跨膜TNF-α单克隆抗体C1可变区基因测序及嵌合抗体的构建[D]. 武汉: 华中科技大学, 2009.
[5] 林春晶, 韦正乙, 蔡勤安, 等. 几种植物转基因表达载体的构建方法[J]. 生物技术, 2008, 18(5): 84-87.
[6] Hartley J L, Temple G F, Brasch M A. DNA cloning using in vitro site-specific recombination[J]. Genome Research, 2000, 10(11): 1 788-1 795.
[7] Katzen F. GatewayR recombinational cloning: a biological operating system[J]. Expert Opinion Drug Discovery, 2007, 2(4): 571-589.
[8] Karimi M, Depicker A, Hilson P. Recombinational cloning with plant gateway vectors[J]. Plant Physiology, 2007, 145(4): 1 144-1 154.
[9] Joubès J, De Schutter K, Verkest A, et al. Conditional, recombinase-mediated expression of genes in plant cell cultures[J]. The Plant Journal, 2004, 37(6): 889-896.
[10] Landy A. Dynamic, structural, and regulatory aspects of lambda site-specific recombination[J]. Annual Review of Biochemistry, 1989, 58(1): 913-941.
[11] Bushman W, Thompson J F, Vargas L, et al. Control of directionality in lambda site specific recombination[J]. Science (New York, NY), 1985, 230(4 728): 906.
[12] Cheo D L, Titus S A, Byrd D R N, et al. Concerted assembly and cloning of multiple DNA segments using in vitro site-specific recombination: functional analysis of multi-segment expression clones[J]. Genome Research, 2004, 14(10B): 2 111-2 120.
[13] Earley K W, Haag J R, Pontes O, et al. Gateway-compatible vectors for plant functional genomics and proteomics[J]. The Plant Journal, 2006, 45(4): 616-629.
[14] Helliwell C, Waterhouse P. Constructs and methods for high-throughput gene silencing in plants[J]. Methods, 2003, 30(4): 289-295.
[15] Lund B A, Leiros H K S, Bjerga G E K. A high-throughput, restriction-free cloning and screening strategy based on ccdB-gene replacement[J]. Microbial Cell Factories, 2014, 13(1): 38.
[16] 范 睿, 凌 鹏, 顾文亮, 等. 海南蒟 cDNA 文库的构建及评价[J]. 热带作物学报, 2013, 34(4): 636-640.
[17] 潘求真, 徐曙光, 李 佳, 等. 绿色荧光蛋白表达载体的改造和表达[J]. 黑龙江畜牧兽医, 2007(1): 13-15.
[18] 周 红, 姜良良, 燕 飞, 等. 一种无标记基因植物表达载体的改造方法[J]. 浙江农业学报, 2013, 25(4): 804-807.
[19] 苏 宁, 孙 萌, 李轶女, 等. 水稻叶绿体16S启动子克隆改造、 载体构建及转化研究[J]. 植物学报, 2003, 20(3): 295-301.
[20] 李敬涛, 孙新华, 余 刚, 等. 几种基于pCAMBIA系列多用途新型植物表达载体的改建及优化[J]. 吉林大学学报: 理学版, 2014, 52(2): 371-375.
[21] 李 磊, 薛 芗, 左示敏, 等. 一种改造载体多克隆位点的新方法[J]. 生物技术, 2013, 23(4): 40-43.
[22] 肖素勤, 孙 振, 轩秀霞, 等. 用于通路(Gateway)克隆技术的植物表达载体研究进展[J]. 植物科学学报, 2012, 30(5): 528-544.
[23] 姚玲玲, 王家宁, 黄永章, 等. 利用同尾酶技术构建pET15b-PEP-1-CAT重组质粒[J]. 郧阳医学院学报, 2006, 25(1): 1-6.
[24] Parr R D, Ball J M. New donor vector for generation of histidine-tagged fusion proteins using the Gateway Cloning System[J]. Plasmid, 2003, 49(2): 179-183.
猜你喜欢
中国中药杂志(2017年2期)2017-03-25
江苏农业科学(2016年10期)2017-02-05
江苏农业科学(2016年7期)2016-10-20
江苏农业科学(2016年7期)2016-10-20
湖南大学学报·自然科学版(2016年6期)2016-07-14
江苏农业科学(2016年4期)2016-06-14
江苏农业科学(2015年9期)2015-10-20
热带农业科学(2015年3期)2015-04-28
山东农业科学(2014年1期)2015-03-09
山东农业科学(2014年9期)2015-01-07
