
焙烧温度对CeCu氧化物催化剂上苯基挥发性有机物催化燃烧的影响
2015-05-14 09:27杜琴香周桂林
石油化工 2015年8期
杜琴香,周桂林,杨 瑶
(重庆工商大学 材料科学与工程系 催化与功能有机分子重庆市重点实验室废油资源化技术与装备教育部工程研究中心,重庆 400067)
石油化工生产过程中排放出低浓度的苯基挥发性有机物(PVOCs)是大气污染的重要来源。随着大气环境的不断恶化和人们对环境保护意识的提高,有效地去除大气中微量PVOCs受到了广泛关注。目前,国内外对挥发性有机物(VOCs)主要采用吸收、吸附、冷凝、膜分离、等离子体、生物处理、直接燃烧和催化燃烧等方法处理。催化燃烧是发生在催化剂表面的完全氧化反应,可实现石油化工工业尾气中微量PVOCs在温和的条件下进行无焰燃烧,并生成CO2和H2O。因此,催化燃烧方法成为了处理石油化工工业尾气中微量PVOCs研究的热点。
催化燃烧法的关键是选择合适的催化材料,贵金属(如Pt,Pd,Ag,Au等)催化剂可在低温下实现气体中微量VOCs的催化燃烧[1-4]。但是贵金属催化剂存在易烧结及中毒、价格昂贵、资源短缺等缺点,限制了其广泛应用。非贵金属氧化物催化剂具有价格低廉、资源丰富等优点。对非贵金属氧化物催化剂的研究发现,复合氧化物催化剂对甲苯具有良好的催化燃烧活性,但存在热稳定差、结构不稳定且活性组分易流失等缺点[5-7]。针对非贵金属氧化物催化剂的缺点,人们进一步开展了复合氧化物催化剂的研究。CeO2或Ce基氧化物催化剂具有独特的催化氧化和还原性质及热稳定性能,且向CeO2晶格中引入其他过渡金属离子能有效地改善其氧化还原性和储氧释氧性能[8-10],CeCu氧化物催化剂表现出更优越的PVOCs催化燃烧性能[11-15]。在制备CeCu氧化物催化剂时,焙烧温度是影响其结构和性能的主要因素之一。考察焙烧温度对CeCu氧化物催化剂性能的影响,对制得具有优越PVOCs催化燃烧性能的催化剂具有十分重要的作用。
本工作采用柠檬酸络合法,在不同焙烧温度下制备了3种CeCu氧化物催化剂,采用XRD和H2-TPR方法对其进行表征,考察焙烧温度对CeCu氧化物催化剂催化PVOCs燃烧活性的影响。
1 实验部分
1.1 催化剂的制备方法
采用柠檬酸络合法制备CeCu氧化物催化剂。按n(Ce)∶n(Cu)=3.0、n(柠檬酸)∶n(Ce+Cu)=1.8的比例分别称取定量的硝酸铜、硝酸铈铵和柠檬酸,将其溶于蒸馏水中,并在80 ℃恒温搅拌下蒸干。将得到的固体物置于烘箱中于100 ℃下干燥过夜。干燥后的固体物,分别在300,350,400 ℃下焙烧3 h制得相应的CeCu氧化物催化剂,分别记作CeCu(300),CeCu(350),CeCu(400)。
1.2 催化剂的表征方法
采用日本理学株式会社Rigaku D/Max-2500/PC型X射线衍射仪进行XRD表征。分析条件:Cu Kα射线,Ni滤波,管电压40 kV,管电流200 mA,扫描速率为5 (°)/min,扫描区间2θ=20°~80°,扫描步长为0.02°。
H2-TPR表征:将30 mg催化剂装入U形石英反应管中,以氢气含量为5.0 %(φ)的H2-Ar混合气为还原气,还原气体的流量经流量计控制在25 mL/min。待记录基线平稳后,以10 K/min的速率由室温升温到设定温度,由TCD检测还原过程中耗氢量并记录TPR谱图。
1.3 催化剂的评价方法
采用固定床反应器评价CeCu氧化物催化剂对PVOCs(苯、甲苯、乙苯和二甲苯)的催化燃烧活性。称取50 mg催化剂装入固定床反应器的恒温段,热电偶传感器置于催化剂床层中间,通过程序升温控制仪控制催化剂床层的温度。在空气气氛中反应器内温度达到设定温度后,切换加入PVOCs含量为1.0%(φ)的混合空气。通过流量计控制空气流量,并控制反应气空速为66 L/(g·h)。
采用上海精密科学仪器有限公司GC1022型气相色谱仪在线分析反应前后混合气中PVOCs的含量,以PVOCs或甲苯的转化率为指标评价CeCu氧化物催化剂的燃烧活性。由式(1)计算转化率(X)。

式中,φin为原料中PVOCs的体积分数,%;φout为尾气中PVOCs的体积分数,%。
2 结果与讨论
2.1 XRD表征结果
不同焙烧温度的CeCu氧化物催化剂的XRD谱图见图1。由图1可知,CeCu氧化物催化剂在2θ=28.5°,33.1°,47.5°,56.3°,59.1°,69.4°,76.7°处出现明显的衍射峰,这些衍射峰与萤石结构的立方晶型CeO2的特征衍射峰相似,且衍射峰强度随催化剂焙烧温度的升高而增强;在2θ=35.56°,38.68°处出现弱的衍射峰,为CuO的特征衍射峰。

图1 不同焙烧温度的CeCu氧化物催化剂的XRD谱图Fig.1 XRD spectra of the CeCu oxide catalysts calcined at different temperature.
由XRD表征结果可知,CeCu氧化物主要以CeCu氧化物固溶体的形式存在。CeCu氧化物催化剂按Cu/(Ce+Cu)摩尔比为0.25制备,即该系列催化体系中含有大量的Cu物种,但CuO晶相的XRD衍射峰比较弱。这可归因于大量的CuO物种溶入到CeO2晶格中,形成了CeCu氧化物固溶体;或部分CuO物种以高分散或无定形态存在于催化剂中。
CeCu氧化物催化剂通过络合法制得,所使用的柠檬酸络合剂能与金属离子(Cu2+或Ce4+)形成络合物,使得Ce和Cu原子能均匀混合,为Cu物种有效地溶入到CeO2晶格中提供了有利条件。因此,CuO物种主要以CeCu氧化物固溶体的形式存在于CeCu氧化物催化体系中。CeCu氧化物固溶体的形成可使CuO与CeO2之间产生强相互作用,使其具有的性质与单组分CuO和CeO2之间存在明显的差异性[16]。一方面,CuO与CeO2之间产生强相互作用力削弱了Cu—O键和Ce—O键,导致其在反应中更易断裂产生高活性的氧物种;另一方面,CeCu氧化物固溶体中存在的Cu+/Cu2+与Ce3+/Ce4+离子对能确保催化体系中电子和氧离子的迁移及活性位的快速再生[17]。
由图1可知,不同焙烧温度下得到的CeCu氧化物催化剂均出现了较弱的CuO晶相衍射峰,表明CeCu氧化物催化剂中有少量的CuO晶相形成,这可归因于催化体系中CuO物种含量超过了CeO2晶格的饱和溶量,导致在焙烧过程中部分CuO聚集形成了CuO晶相;随焙烧温度的升高,CeCu氧化物固溶体的衍射峰强弱顺序为:CeCu(400)>CeCu(350)> CeCu(300),表明焙烧温度的升高提高了CeCu氧化物固溶体的结晶度。同时,焙烧温度的升高会引起催化体系中物种的聚集,增大催化剂的晶粒度。催化剂的活性会受到催化剂晶粒度大小的影响,催化剂的晶粒度越小,晶格畸变率就越大,越有利于提高催化剂的燃烧活性[18]。
硝酸铜完全分解温度为310 ℃[19],硝酸铈铵完全分解温度为290 ℃[20],即当焙烧温度为300 ℃时, CeCu氧化物催化体系中可能会有部分硝酸盐不能完全分解,且低的焙烧温度不利于CeCu氧化物固溶体的形成,而较高的焙烧温度(400℃)会削弱催化剂的燃烧活性。因此,制备CeCu氧化物催化剂适宜的焙烧温度为350 ℃。
2.2 H2-TPR表征结果
不同温度焙烧的CeCu氧化物催化剂的H2-TPR曲线见图2。由图2可知,当还原温度为125 ℃时,H2-TPR曲线均明显偏离基线;随还原温度的升高,在150~250 ℃范围内出现两个明显的α和β耗氢峰,且β耗氢峰的峰强度和峰面积均大于α耗氢峰;CeCu(350)氧化物催化剂的β耗氢峰对应的还原温度低于CeCu(300)和CeCu(400)氧化物催化剂;随焙烧温度的升高,CeCu氧化物催化剂的α耗氢峰所对应的还原温度降低。

图2 不同温度焙烧的CeCu氧化物催化剂的H2-TPR曲线Fig.2 H2-TPR curves of the CeCu oxide catalysts calcined at different temperature.
纯相CeO2在500 ℃和800 ℃时可形成明显的耗氢峰,其分别对应于表面高分散CeO2的还原和体相聚集态CeO2的还原[21-22];纯相CuO在300 ℃时能被还原形成耗氢峰[23]。由图2可知,CeCu氧化物催化剂的还原温度明显低于纯相CeO2和CuO的还原温度。由XRD分析结果可知,Cu物种能很好地溶入到CeO2晶格中形成CeCu氧化物固溶体,CeCu氧化物固溶体中CuO与CeO2间的强相互作用能有效地削弱“金属—氧”键,从而提高金属氧化物的可还原性能,使得CeCu氧化物催化剂具有良好的低温还原性。
图2中α耗氢峰可归属于CeCu氧化物催化剂表面高分散态且与CeO2有强相互作用的CuO物种的还原;β耗氢峰可归属于CeCu氧化物催化剂中CeCu氧化物固溶体的还原。
在α耗氢峰区域,随焙烧温度的升高,CeCu氧化物催化剂中CuO物种的低温可还原性增强,这归因于焙烧温度升高增强了CuO与CeO2的相互作用。
在β耗氢峰区域,CeCu(350)氧化物催化剂的还原温度低于CeCu(300)和CeCu(400)氧化物催化剂,可归因于焙烧温度的升高促使Cu物种充分地溶入CeO2晶格,同时也促进了CeCu氧化物固溶体中CuO与CeO2间的相互作用,进一步削弱了“金属—氧”键,提高了CeCu氧化物固溶体的可还原性,从而导致CeCu(350)氧化物催化剂的可还原性能优于CeCu(300)氧化物催化剂;但随焙烧温度的继续升高,CeCu氧化物固溶体的结晶度也会提高,通常高结晶度的CeCu氧化物固溶体更难还原,故CeCu(350)氧化物固溶体的还原性能优于CeCu(400)氧化物固溶体。
CeCu氧化物固溶体可为催化氧化或燃烧反应提供大量的高活性氧物种,使得CeCu氧化物催化剂具有高的催化氧化或燃烧活性。结合H2-TPR表征结果可知,CeCu(350)氧化物催化剂更易在温和的条件下提供高活性氧物种,故该催化剂可能会表现出优越的催化氧化或燃烧反应性能。
2.3 焙烧温度对CeCu氧化物催化剂燃烧活性的影响
焙烧温度对CeCu氧化物催化剂上甲苯催化燃烧活性的影响见图3。
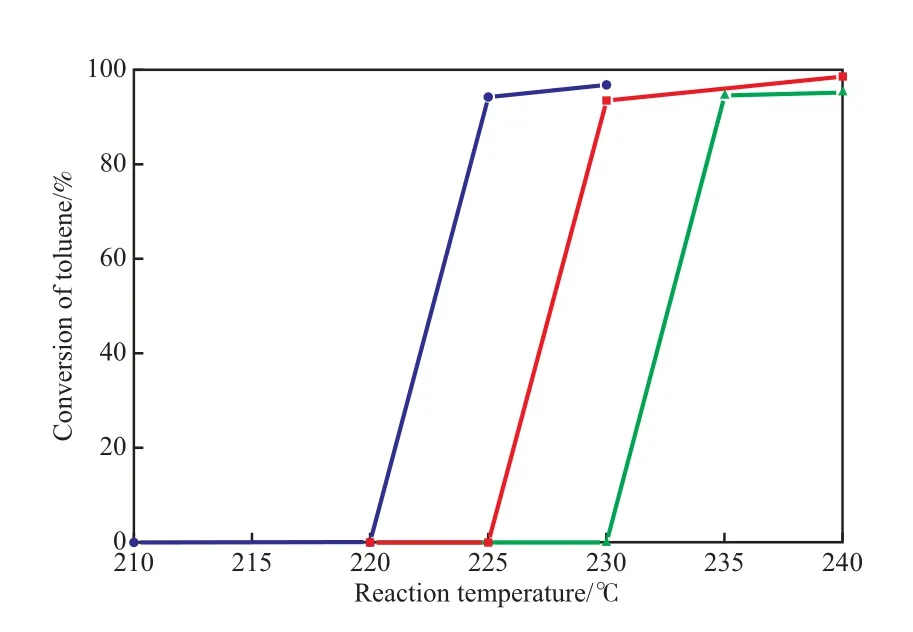
图3 焙烧温度对CeCu氧化物催化剂上甲苯催化燃烧活性的影响Fig.3 Effects of calcination temperature on the activities of the CeCu oxide catalysts in the catalytic combustion of toluene.
由图3可知,CeCu氧化物催化剂对甲苯具有良好的催化燃烧活性; CeCu(350)氧化物催化剂对甲苯表现出最佳的催化燃烧活性,起始反应温度仅为220 ℃,当反应温度为225 ℃和230 ℃时,甲苯的转化率分别达到94.3%和96.8%;CeCu(400)氧化物催化剂对甲苯的催化燃烧活性低于CeCu(350)氧化物催化剂,在反应温度为230 ℃和240 ℃时,甲苯的转化率分别为93.5%和98.6%;CeCu(300)氧化物催化剂上甲苯的催化燃烧活性最低,当反应温度为235 ℃和240 ℃时,甲苯的转化率分别为94.6%和95.3%。
甲苯在空气中直接燃烧温度高达535 ℃。在CeCu氧化物催化剂作用下,大幅降低了甲苯的燃烧温度,即CeCu氧化物催化剂显著提高了甲苯的低温可燃性。由于CeCu氧化物催化剂中存在CeCu氧化物固溶体,CeCu氧化物固溶体中CeO2与CuO间的强相互作用削弱了“Cu—O”和“Ce—O”键,使其更易断裂产生高活性氧物种。同时,Cu物种溶入到CeO2晶格中,促进了催化剂中大量氧空穴的形成,大量的氧空穴不仅为氧分子的吸附和活化提供活性中心,还能提高晶格氧在催化剂中的转移和扩散速率,也能有效地提高催化剂活化氧分子的能力,从而能提高催化剂提供高活性氧物种的能力[24]。H2-TPR表征结果证实,CeCu氧化物催化剂可为氧化反应提供大量的活性氧物种,能为甲苯在CeCu氧化物催化剂上高效燃烧提供物质保证。因此,CeCu氧化物催化剂表现出了高的甲苯低温催化燃烧活性。
CeCu氧化物催化剂对甲苯的催化燃烧活性高低的顺序为:CeCu(350)> CeCu(400)> CeCu(300)。这归因于低的焙烧温度(300 ℃)不利于CuO物种有效地溶入到CeO2晶格中而促进CeO2与CuO间的强相互作用的产生[21,25],进而达不到充分活化催化剂的目的,以至于不能形成足够的高活性氧物种;过高的焙烧温度(400 ℃)能有效地提高催化剂的结晶度,高的结晶度会削弱催化剂提供活性氧物种的能力;适宜的焙烧温度(350 ℃)有利于CeO2与CuO间形成强的相互作用,从而促进活性氧物种的形成,以至于提高催化剂的催化氧化或燃烧性能。
CeCu(350)氧化物催化剂的低温可还原性能优于CeCu(300)和CeCu(400)氧化物催化剂,表明CeCu350氧化物催化剂在温和条件下更易为甲苯分子催化燃烧提供活性氧物种,从而促进甲苯分子在该催化剂上进行低温催化燃烧反应。因此,CeCu(350)氧化物催化剂对甲苯的催化燃烧活性优于CeCu(300)和CeCu(400)氧化物催化剂,且CeCu(400)氧化物催化剂的燃烧活性优于CeCu(300)氧化物催化剂。
2.4 CeCu(350)氧化物催化剂对PVOCs的催化燃烧活性
CeCu(350)氧化物催化剂上PVOCs的转化率见图4。由图4可知,当反应温度为205 ℃和210 ℃时,乙苯的转化率分别高达99.0%和99.4%;当反应温度为220 ℃和230 ℃时,二甲苯的转化率分别达到98.9%和98.6%;当反应温度为225 ℃和230 ℃时,苯的转化率分别为62.8%和62.5%,甲苯的转化率分别达到94.3%和96.8%。

图4 CeCu(350)氧化物催化剂上PVOCs的转化率Fig.4 Conversions of volatile phenyl compounds(PVOCs)over the CeCu(350)oxide catalyst.
在CeCu(350)氧化物催化剂上PVOCs催化燃烧活性存在明显的差异,这是由于PVOCs中不同物质自身性质不同所引起的。苯、甲苯、邻二甲苯和乙苯的偶极矩分别为0,0.36,0.62,0.59 D(D =3.334×10-30C·m)[26]。分子极性大小的顺序为:二甲苯>乙苯>甲苯>苯。CeCu(350)氧化物催化剂表面存在大量的“金属—氧”键,致使该催化剂具备强极性表面,从而有利于极性大的有机分子在催化剂表面的吸附和活化。因此,CeCu(350)氧化物催化剂吸附和活化二甲苯的能力最强,其次为乙苯、甲苯、苯。苯分子具有高度对称的芳香环结构,其结构非常稳定,甲苯、二甲苯或乙苯上的取代基,影响苯环结构的稳定性和极性。因此,甲苯、二甲苯和乙苯比苯更易发生氧化反应。
甲苯和二甲苯因苯环上甲基取代基的个数不同,从而导致二甲苯的苯环结构的稳定性小于甲苯;同时,二甲苯的分子极性大于甲苯,CeCu(350)氧化物催化剂对二甲苯的催化燃烧活性明显高于甲苯。
二甲苯和乙苯分子因苯环上取代基的不同导致分子性质不同,二甲苯比乙苯具有更大的分子尺寸[27],空间位阻效应降低了催化剂对二甲苯分子的吸附性能;二甲苯比乙苯分子具有更大的磁化率[26],二甲苯分子在CeCu(350)氧化物催化剂表面难以脱附,这可能会阻碍催化剂对O2分子的吸附,从而降低了催化剂提供活性氧物种的能力,因此CeCu(350)氧化物催化剂对乙苯催化燃烧的活性比二甲苯高。
综上所述,苯、甲苯、二甲苯和乙苯在CeCu(350)氧化物催化剂上的催化燃烧活性高低的顺序为:乙苯>二甲苯>甲苯>苯。
2.5 CeCu(350)氧化物催化剂的稳定性
CeCu(350)氧化物催化剂上甲苯催化燃烧的稳定性见图5。由图5可见,对于氧化性气氛(空气99.0%(φ)-甲苯1.0%(φ)),在220 ℃时CeCu(350)氧化催化剂对甲苯的转化率在9 min内从33.7%逐渐降到了0.1%;在225 ℃时,CeCu(350)氧化物催化剂对甲苯的转化率超过93%,且在800 min内未出现明显的失活;对于惰性气氛(N299.0%(φ)-甲苯1.0%(φ)),在225 ℃时,CeCu(350)氧化物催化剂上甲苯转化率在15 min内从64.3%逐渐降到0.1%以下。
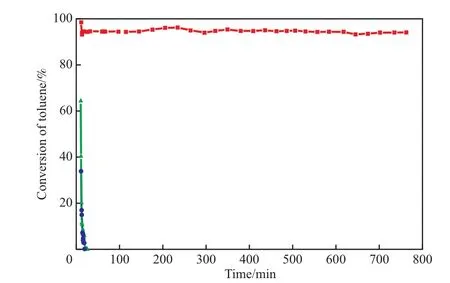
图5 CeCu(350)氧化物催化剂上甲苯的转化率与反应时间的关系Fig.5 Relationship between reaction time and the catalytic combustion conversion of toluene on the CeCu(350)catalyst.
由图5可知,反应温度和气氛对甲苯的转化率有明显的影响。在220 ℃时,CeCu(350)氧化物催化剂对甲苯分子的起始转化率为33.7%,9 min之后降至0.1%,可因归于CeCu(350)氧化物催化剂对甲苯分子的吸附,催化剂对甲苯分子吸附饱和后不再表现出甲苯的转化。在空气和甲苯组成的氧化性气氛中,反应温度为225 ℃时,CeCu(350)氧化物催化剂能有效地活化氧分子形成高活性氧物种,为甲苯分子在CeCu(350)氧化物催化剂上高效燃烧提供物质保证,以至于CeCu(350)氧化物催化剂表现出高的甲苯分子的催化燃烧活性和良好的使用稳定性。
在N2和甲苯组成的惰性气氛中,反应温度为225 ℃时,甲苯的转化率可达64.3%。这是由于CeCu(350)氧化物催化剂表面存在大量活性氧物种参与了燃烧反应以及甲苯在催化剂上的吸附,使得在反应初始阶段甲苯的转化率高达64.3%;随着反应的进行,活性氧物种被消耗及催化剂对甲苯的吸附达到平衡,导致甲苯的转化率在15 min内急剧降至0.1%。
综上所述,CeCu(350)氧化物催化剂在一定条件下能有效地活化氧分子,为甲苯的催化燃烧反应提供高活性氧物种,从而使得甲苯能高效地实现燃烧反应并表现出良好的催化稳定性。
3 结论
1) 采用柠檬酸络合法,改变焙烧温度制备了3种CeCu(300),CeCu(350),CeCu(400)氧化物催化剂。焙烧温度升高有利于形成CeCu氧化物固溶体,也可使固溶体的结晶度提高。与CeCu(300)和CeCu(400)氧化物催化剂相比,CeCu(350)氧化物催化剂具有适宜的结晶度、较好的可还原性。
2)CeCu氧化物催化剂上甲苯的催化燃烧活性高低的顺序为:CeCu(350)>CeCu(400)> CeCu(300)。当反应温度为225 ℃时,CeCu(350)催化剂上甲苯转化率高达94.3%。
3)CeCu(350)氧化物催化剂上PVOCs转化率高低的顺序为:乙苯>二甲苯>甲苯>苯。
4)在225 ℃下,800 mi n内,CeCu(350)氧化物催化剂上甲苯的转化率达93%以上,CeCu(350)氧化物催化剂对甲苯催化燃烧具有良好的稳定性。
[1]Ordóñez S,Bello L,Sastre H,et al. Kinetics of the Deep Oxidation of Benzene,Toluene,n-Hexane and Their Binary Mixtures over a Platinum on γ-Alumina Catalyst[J]. Appl Catal,B,2002,38(2):139 - 149.
[2]邱健斌,洪惠云,刘云珍. Pd-CeZr/SiC催化剂的制备及其催化氧化性能[J]. 石油化工,2014,43(2):150 - 154.
[3]Scire S,Minico S,Crisafulli C,et al. Catalytic Combustion of Volatile Organic Compounds on Gold/Cerium Oxide Catalysts[J]. Appl Catal,B,2003,40(1):43 - 49.
[4]王路存,苏方正,黄新松. 高性能纳米金催化剂的研究进展[J]. 石油化工,2007,36(9):869 - 875.
[5]Li W B,Chu W B,Zhuang M,et al. Catalytic Oxidation of Toluene on Mn-Containing Mixed Oxides Prepared in Reverse Microemulsions[J]. Catal Today,2004,93/95:205 - 209.
[6]李永峰,李宇,余林,等. 锰掺杂六铝酸镧催化剂上甲苯催化燃烧净化研究[J]. 燃料化学学报,2012,40(1):100 -104.
[7]任倩茹,张泽凯,徐彪,等. 负载型钙钛矿La0.8Sr0.2MnO3/SBA-15催化燃烧甲苯的研究 [J]. 燃料化学学报,2012,40(9):1142 - 1146.
[8]Usmen R K,Graham G W,Watkins W L,et al. Incorporation of La3+into a Pt/CeO2/Al2O3Catalyst[J]. Catal Lett,1994,30(1/4):53 - 63.
[9]Bensalem A,Bozon-Verduraz F,Delamar M,et al. Preparation and Characterization of Highly Dispersed Silica-Supported Ceria[J]. Appl Catal,A,1995,121(1):81 - 93.
[10]Zamar F,Trovarelli A,Leitenburg C de,et al. CeO2-Based Solid Solutions with the Fluorite Structure as Novel and Effective Catalysts for Methane Combustion[J]. J Chem Soc Chem Commun,1995(9):965 - 966.
[11]Zhou Guilin,Lan Han,Yang Xiaoqing,et al. Effects of the Structure of Ce-Cu Catalysts on the Catalytic Combustion of Toluene in Air[J]. Ceram Int,2013,39(4):3677 - 3683.
[12]Shan Wenjuan,Feng Zhaochi,Li Zhonglai,et al. Oxidative Steam Reforming of Methanol on Ce0.9Cu0.1OyCatalysts Prepared by Deposition-Precipitation,Coprecipitation,and Complexation-Combustion Methods[J]. J Catal,2004,228(1):206 - 217.
[13]李孔斋,王华,魏永刚,等. 铈基复合氧化物中晶格氧用于甲烷部分氧化制合成气[J]. 燃料化学学报,2008,36(1):83 - 88.
[14]Zhou Guilin,Lan Han,Gao Taotao,et al. Influence of Ce/Cu Ratio on the Performance of Ordered Mesoporous CeCu Composite Oxide Catalysts[J]. Chem Eng J,2014,246:53 - 63.
[15]杜琴香,周桂林,王慧,等. Ce0.75Cu0.25-xO2固溶体催化剂用于PVOCs催化燃烧[J]. 工业催化,2014,22(2):123 - 127.
[16]Luo Mengfei,Ma Jingmeng,Lu Jiqing,et al. High-Surface Area CuO-CeO2Catalysts Prepared by a Surfactant-Templated Method for Low-Temperature CO Oxidation[J]. J Catal,2007,246(1):52 - 59.
[17]Menon U,Poelman H,Bliznuk V,et al. Nature of the Active Sites for the Total Oxidation of Toluene by CuOCeO2/Al2O3[J].J Catal,2012,295:91 - 103.
[18]刘成文. Ce1-xFexO2复合氧化物的制备及其甲烷催化燃烧性能的研究[D]. 南昌:南昌大学理学院化学系,2007.
[19]Ghose J,Kanungo A. Studies on the Thermal Decomposition of Cu(NO3)2·3H2O[J]. J Therm Anal,1981,20:459 - 462.
[20]Pokol G,Leskelä T,Niinistö L. Thermal Behavior of Sulphato and Nitrato Complexes of Cerium(Ⅳ)[J]. J Therm Anal,1994,42:343 - 359.
[21]Avgouropoulos G,Ioannides T. Effect of Synthesis Parameters on Catalytic Properties of CuO-CeO2[J]. Appl Catal,B,2006,67(1):1 - 11.
[22]Zeng Jia,Zhou Guilin,Ai Yongmei,et al. Catalytic Wet Peroxide Oxidation of Chlorophenol over a Ce0.86Cu0.14-xO[J].Int J Chem React Eng,2013,11(1):577 - 585.
[23]Djinović P,Batista J,Pintar A. Calcination Temperature and CuO Loading Dependence on CuO-CeO2Catalyst Activity for Water-Gas Shift Reaction[J]. Appl Catal,A,2008,347(1):23 - 33.
[24]Rodriguez J A,Hanson J C,Kim J Y,et al. Properties of CeO2and Ce1-xZrxO2Nanoparticles:X-Ray Absorption Near-Edge Spectrosc opy, Density Functional and Time-Resolved X-Ray Diffraction Studies[J]. J Phys Chem B,2003,107(15):3535 - 3543.
[25]Avgouropoulos G,Ioannides T,Matralis H. Influence of the Preparation Method on the Performance of CuO-CeO2Catalysts for the Selective Oxidation of CO[J]. Appl Catal,B,2005,56(1):87 - 93.
[26]姚允斌,解涛,高英敏. 物理化学手册[M]. 上海:科学技术出版社,1985:227.
[27]Raj K,Malar E J,Vijayaraghavan V R. Shape-Selective Reaction with AEL and AFI Type Molecular Sieves Alkylation of Benzene,Toluene and Ethylbenzene with Ethanol,2-Propanol,Methanol,and t-Butanol[J]. J Mol Catal A:Chem,2006,243(1):99 - 105.
猜你喜欢
建材发展导向(2020年15期)2020-11-26
工业催化(2020年5期)2020-06-23
化工管理(2020年11期)2020-04-23
四川水泥(2019年9期)2019-02-16
中国医学装备(2016年11期)2016-12-09
西南国防医药(2016年6期)2016-12-01
现代检验医学杂志(2016年1期)2016-11-12
山东医药(2015年38期)2015-12-07
华东理工大学学报(自然科学版)(2015年3期)2015-11-07
医学研究杂志(2015年4期)2015-06-10
