
2,5-二溴-4-硝基咪唑的合成*
2015-04-23 10:55倪珊梅胡金辉顾庆美周琢强
合成化学 2015年3期
倪珊梅,胡金辉,顾庆美,刘 凯,周琢强
(华南农业大学理学院,广东 广州 510642)
2,5-二溴-4-硝基咪唑(2)是一种重要的药物中间体,但近年来关于其合成的研究报道较少。2012年,Kim Pil Ho等[1]以 4-硝基咪唑为原料,碳酸氢钠为催化剂,溴素为溴代试剂,水为溶剂,于40℃反应12 h合成了2,收率67%;2013年,Pedada Srinivasa Rao等[2]改良了上述工艺,在固定其他反应工艺不变的条件下,于0℃反应5 h后升温至65℃继续反应6 h,收率提高至88%。
本课题组多次重复文献[2]方法后发现,效果不佳,其原因可能是溴素溶于水发生歧化反应生成的氢溴酸和次溴酸与碳酸氢钠中和,致使溴素失效。此外,反应时间太长(均>10 h),反应试剂危害环境等弊端也不利于工业化生产。
为此,本文设计了以咪唑为原料,经硝化和溴化反应合成2(Scheme 1)的合成路线,总收率80.3%,其结构经1H NMR和IR确认。该合成路线工艺简单,易于操作,有一定的市场应用价值。
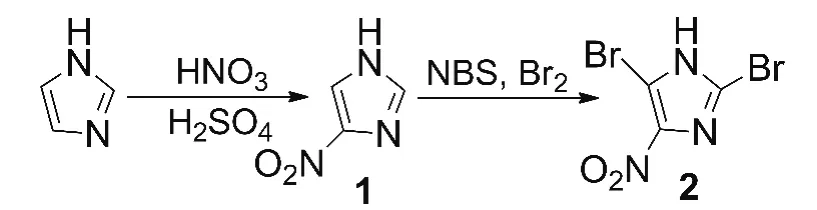
Scheme 1
1 实验部分
1.1 仪器与试剂
X-4型熔点仪(温度未校正);Bruker-AVANCE III 600 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Nicolet AVA7AR360-FT型红外光谱仪(KBr压片)。
咪唑使用前经纯化处理(加入无水乙醇,回流,趁热抽滤,滤液冰冻2 h;抽滤,滤饼于40℃用P2O5干燥2 h);醋酸使用前经无水处理(加入乙酸酐和三氧化铬,回流 3 h,常压蒸馏,收集117℃ ~118℃馏分);N-溴代琥珀酰亚胺(NBS)使用前经纯化处理(加入热水,加热使其完全溶解;冰冻,抽滤,滤饼于50℃用P2O5干燥4 h);其余所用试剂均为分析纯。
1.2 合成
(1)4-硝基咪唑(1)的合成
在干燥的单口烧瓶中加入咪唑1.70 g(25 mmol),缓慢加入冰冻浓硫酸,加毕,搅拌使其溶解;冰浴冷却,缓慢滴加90%浓硝酸2.63 g(37.5 mmol),滴毕,于50℃反应8 h。倒入50 mL冰水中,待气泡消失,抽滤,滤饼依次用冰水(2×15 mL)和乙醇(2×15 mL)洗涤,于50℃用P2O5干燥2 h得淡黄色固体 1 2.58 g,收率 91.3%,m.p.308 ℃ ~ 310 ℃ (309 ℃ ~ 310 ℃[3]);1H NMR δ:13.23(s,1H),8.31(d,J=1.3 Hz,1H),7.84(d,J=1.2 Hz,1H);IR ν:3 439,3 141,2 822,1 556,1 510,1 497,1 434,1 382,1 253,991,867 cm-1。
(2)2的合成
在干燥的三口烧瓶中依次加入1 1.13 g(10 mmol),醋酸40 mL,铁粉60 mg(1 mmol)和溴素240 mg(1.5 mmol),搅拌下于室温分批加入NBS 3.74 g(21 mmol),加毕,回流(118 ℃)反应 4 h。冷却至室温,常压蒸除大部分醋酸,残留液倒入100 mL冰水中,搅拌10 min。抽滤,滤饼用冰水(3×50 mL)洗涤,加入乙酸乙酯100 mL溶解,有机层依次用饱和碳酸氢钠溶液(2×30 mL)和饱和食盐水(3×30 mL)洗涤(维持有机层pH 7左右),用无水硫酸钠干燥,浓缩后经硅胶柱层析[洗脱剂A:V(石油醚)∶V(乙酸乙酯)=8∶1]纯化得类白色固体 2 2.38 g,收率 88.0%,f.p.193℃,b.p.396 ℃ ~397 ℃(396 ℃[4]);1H NMR δ:14.12(s,1H,NH);IR ν:3 448,2 921,1 655,1 533,1 395,1 296,1 178,981,656 cm-1。
2 结果与讨论
2.1 合成
(1)1的合成
用混酸硝化咪唑,不同加料方式得到的产物不同[5]。采用反加法加料,即在混酸中加入咪唑,体系中硝鎓离子浓度始终大于咪唑浓度,生成的1或5-硝基咪唑可被进一步硝化,咪唑2-位、4-位或5-位氢均有可能被硝基取代,导致副产物增多,反应不可控制。为避免生成多取代产物,实验中选用正加法加料,即先用浓硫酸溶解咪唑,再缓慢滴加发烟硝酸。
溶解咪唑所用浓硫酸需先冷冻,摇荡下缓慢加入。由于浓硫酸溶解咪唑过程中会产生大量热,并释放大量白烟,若加入浓硫酸速度过快,会发生爆沸甚至爆炸。因此,浓硫酸的加入速度必须很慢,直至咪唑全部溶解。
滴加发烟硝酸时,由于反应剧烈放热,为防止体系爆沸,实验中采用了冰浴冷却,缓慢滴加发烟硝酸的方式,滴毕,于50℃反应8 h。反应温度太低,即便搅拌20 h,反应仍不完全;温度过高,容易生成多硝基取代物,副产物增多,50℃为咪唑4-位引入硝基的最佳温度。
(2)2的合成
2的合成是本路线的关键。为提高2收率,考察了溴代试剂、催化剂、溶剂和反应温度对2收率的影响,结果见表1。
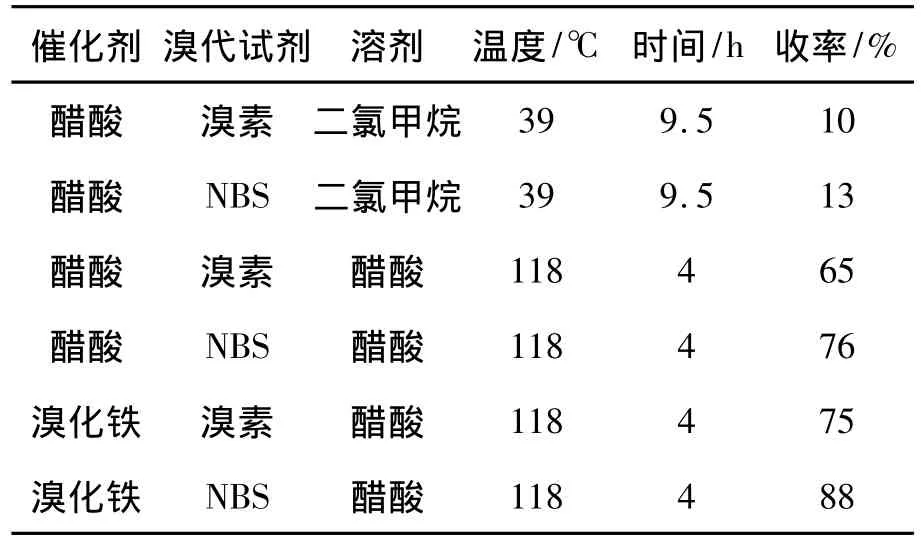
表1 反应条件对2收率的影响*Table 1 The effects of reaction conditions on the yield of 2
由表1可见,低温下使用二氯甲烷作溶剂,2收率较低。其可能原因是,1受其硝基取代基的强致钝效应影响,反应活性降低,无法有效引发反应。以NBS作溴代试剂,2收率较高,其原因可能是,NBS在醋酸作用下比溴素更易极化,生成溴正离子,对咪唑环4-位的亲电进攻更为有效。醋酸为溶剂时,溴素和NBS对2收率影响差异更为明显,其原因可能在于NBS异裂在达到引发温度后便自发进行,而溴素在回流时易挥发,导致2收率不高。溴化铁比醋酸能够更有效地催化反应。此外,还开展了以氯化铝或氯化锌为催化剂的对照实验,催化效果均不及溴化铁,其可能原因是,氯化铝和氯化锌吸水严重,导致催化失效,而氯化铝、氯化锌无水纯化均需在HCl氛围下升华,实验室条件难以达到。
[1]Kim P H,Kim S H,Lee I Y,et al.Nitroimidazole compound for treating tuberculosis and method for the preparation thereof[P].KR 2 012 060 663,2012.
[2]Pedada S R,Satam V S,Tambade P J,et al.An improved kilogram-scale synthesis of 2-bromo-4-nitro-1H-imidazole:A key building block of nitroimidazole drugs[J].Organic Process Research & Development,2013,17(9):1149-1155.
[3]Suwinski,J,Salwinska E,Watras J,et al.Nitromidazoles.PartⅤ.Chloronitroimidazoles from dinitroimidazoles:A reinvesthgation[J].Polish Journal of Chemistry,1982,56:1261 -1272.
[4]Balaban I E,Pyman F L.Bromo derivatives of glyoxaline[J].Journal of Chemistry Society,Transaction,1922,121:947 -958.
[5]刘慧君,杨林,曹端林.4-硝基咪唑的合成工艺及其热安定性[J].中北大学学报(自然科学版),2006,27(4):331-334.
猜你喜欢
当代化工研究(2022年12期)2022-07-11
中国化工贸易·上旬刊(2020年3期)2020-09-10
——非均布滤饼的局部比阻与平均比阻的测定与计算方法
化工装备技术(2020年4期)2020-09-09
流体机械(2020年5期)2020-06-24
广州化工(2020年11期)2020-03-07
中国司法鉴定(2017年3期)2017-06-24
中学化学(2017年2期)2017-04-01
中学生数理化·中考版(2017年1期)2017-03-29
中国矿业(2017年2期)2017-02-28
广州大学学报(自然科学版)(2015年4期)2015-12-23
