
西方角蝇和截脉角蝇18S rDNA的克隆及其分子系统学研究
2015-04-18 10:41史红蕾杨莲茹杨晓野郑文青
中国兽医杂志 2015年12期
史红蕾,邓 侨,杨莲茹,杨晓野,王 瑞,郑文青,杨 波
(内蒙古农业大学兽医学院农业部动物疾病临床诊疗技术重点实验室,内蒙古呼和浩特010018)
西方角蝇和截脉角蝇18S rDNA的克隆及其分子系统学研究
史红蕾,邓 侨,杨莲茹,杨晓野,王 瑞,郑文青,杨 波
(内蒙古农业大学兽医学院农业部动物疾病临床诊疗技术重点实验室,内蒙古呼和浩特010018)
为了弄清双翅目昆虫西方角蝇(Haematobia.irritans)和截脉角蝇(Haematobia.titillans)18S rDNA基因序列及其分子进化。本研究测定了两种角蝇的18S rDNA基因序列,将其在NCBI中Blast,并将序列与GenBank中已知9种双翅目昆虫的18S rDNA基因序列进行比较分析,找出它们的同源区和多变区,利用全序列和最保守同源区分别构建系统发育树。结果表明,西方角蝇和截脉角蝇18S rDNA基因序列长度均为1 984 bp,二者同源性为96.4%,存在73个识别位点。两种角蝇与GenBank中已登陆西方角蝇的同源性分别为99.1%和95.7%。11种双翅目昆虫18S rDNA基因序列中均有三段保守程度较高的同源区,分别相当于西方角蝇18S rDNA基因序列中320 bp~693 bp,848 bp~1 181 bp和1 606 bp~1 849 bp,其中第一段同源区最为保守。本文在国内外首次报道截脉角蝇18S rDNA基因序列,并证明了西方角蝇18S rDNA基因序列高度保守。利用18S rDNA基因序列中最保守同源区构建的系统发育树,对11种双翅目昆虫分类更符合传统形态学分类结果。
西方角蝇;截脉角蝇;18S rDNA;序列分析
西方角蝇(Haematobia irritans)和截脉角蝇(Haematobia titillans)是内蒙古地区荒漠化草原危害较严重的吸血蝇。它们寄生于骆驼、牛、羊等反刍家畜的体表,以吸血为生,不仅会对家畜造成机械性损伤,还会使其产生强烈应激反应,甚至引起某些虫媒性疾病[1]。赵治国等(2010)研究表明,西方角蝇和截脉角蝇是骆驼斯氏副柔线虫病的传播媒介[2],而骆驼斯氏副柔线虫病是严重危害养驼业的一种寄生虫病。
18S rDNA是指由染色体编码的真核生物核糖体小亚基RNA,是至今发现的DNA序列中最保守的一类[3]。目前,已有许多学者研究昆虫的18S rDNA基因序列,如魏国清等(2006)对鳞翅目昆虫家蚕18S rDNA基因序列的特点及分子系统学进行了研究[4];李海超等(2009)分析了鳞翅目烟夜蛾18S rDNA基因序列[5];王海亭等(2010)克隆了玉米螟的18S rDNA基因序列并做了分子系统学研究[6]。目前,许多生物的18S rDNA基因序列已发表或输入DNA数据库[7-8],为昆虫18S rDNA分子系统学研究奠定了基础。
本研究测定了内蒙古地区西方角蝇和截脉角蝇18S rDNA基因序列,将其在NCBI中Blast,确定两种角蝇的18S rDNA基因序列是否保守,以获得可用于区分两种角蝇的易变区及差异位点;并与GenBank中已知18S rDNA基因序列的9种双翅目昆虫进行比较分析,找出它们的同源区,利用18S rDNA基因序列及其最保守同源区构建系统发育树,以确定两种角蝇在双翅目昆虫中的位置,为双翅目昆虫之间的差异性及其分子系统进化特点研究提供依据。
1 材料与方法
1.1 蝇种来源及基因组DNA提取西方角蝇和截脉角蝇采自内蒙古地区荒漠化草原骆驼生活环境中,分别取其单只胸部肌肉10mg提取基因组DNA。
1.2 引物及PCR反应以王瑛(1999)设计的昆虫18S rDNA基因通用引物[9],对角蝇基因组DNA进行PCR扩增,1、2和3、4两对引物序列如下:
1:5′-TACCCTGGTGTGATCCTGCCAGT-3′;2:5′-ACTAGGGCGGTATCTGATCGC-3′;3:5′-GCGATCAGATACCGCCCTAGT-3′;4:5′-GATCCTTCCT⁃GCAGGTTCACCT-3′。
PCR扩增片段长度为1 086 bp和835 bp。两对引物PCR反应体系均为50μL。反应条件均为94℃预变性10 min,进入循环;94℃变性30 s,58℃退火30 s,72℃延伸1 min,共进行30个循环;72℃延伸10min。
1.3 PCR产物的克隆与测序将PCR胶回收产物与pMD19-TVector连接,把连接产物转入大肠杆菌感受态细胞中,在转化平板上37℃培养12 h,待蓝白斑清晰时,挑选白斑转入含有Amp的LB液体培养基中扩繁,用PCR方法鉴定阳性克隆菌液后测序。
1.4 序列的分析比较利用EditSeq程序对两种角蝇的测序结果进行拼接,获得其18S rDNA基因序列。另在GenBank数据库中获取9种双翅目昆虫的18S rDNA基因序列。利用MegAlign程序处理11种双翅目昆虫18S rDNA基因序列进行排序处理,找出它们的同源区和多变区并构建系统发育树。
2 结果
2.1 测序结果西方角蝇和截脉角蝇18S rDNA基因序列长度均为1 984 bp,分别与GenBank中已登录的西方角蝇18S rDNA基因序列(EU179518.1)同源性为99.1%和95.7%,其中截脉角蝇的18S rDNA基因序列尚未在GenBank中登录。
2.2 同源性比较与序列分析结果11种双翅目昆虫18S rDNA基因序列同源性比较结果见图1,其同源性范围为93.2%~99.3%。通过序列分析可知,11种双翅目昆虫18S rDNA基因具有三段保守程度较高的同源区,分别相当于西方角蝇18S rDNA的320~693 bp、848~1 181 bp、1 569~1 849 bp,其中第一段同源区最为保守。11种双翅目昆虫18S rDNA基因的各碱基含量相近,其A+T含量均高于G+C含量。两种角蝇18S rDNA基因序列之间共有73个识别位点,碱基差异为A与T间的替换。

图1 11种双翅目昆虫18S rDNA基因序列的同源性与差异性比较
2.3 系统发育关系分析结果由11种双翅目昆虫18S rDNA基因序列和最保守同源区分别构建的系统发育树可见(图2、图3),两个系统发育树拓扑结构不一致,虽然都分两个大集落,但结点和进化分支不同。
3 讨论与小结
本研究测定的西方角蝇18S rDNA基因序列与GenBank中已登录的西方角蝇18S rDNA基因序列差异性仅为0.9%,表明西方角蝇18S rDNA基因序列高度保守;而截脉角蝇18S rDNA基因序列属国内外首次报道。选取已知18S rDNA基因序列的9种双翅目昆虫,通过比较它们与两种角蝇18S rDNA基因序列的同源性,可知11种双翅目昆虫18S rDNA基因序列均具有3段保守程度较高的同源区。
上述11种双翅目昆虫分属6科,即蝇科(Mus⁃cidae)、花蝇科(Anthomyiidae)、果蝇科(Drosophili⁃dae)、实蝇科(Tephritidae)、寄蝇科(Tachinidae)和舌蝇科(Glossinoidae)。据中国动物志[10]等资料可知,此六科昆虫均隶属于昆虫纲(Insecta)、双翅目(Diptera)、环裂亚目(Cyclorrhapha)、蝇形支目(Muscomorpha)。蝇形支目昆虫又分为有瓣类和无瓣类,其中蝇科、花蝇科、寄蝇科和舌蝇科属于有瓣类,果蝇科和实蝇科属于无瓣类。本研究以11种双翅目昆虫18S rDNA基因序列和最保守同源区分别构建系统发育树,通过比较发现,由最保守同源区构建的系统发育树的拓扑结构反映的种系关系更符合传统形态学分类结果,与传统形态学蝇形支目的物种分类基本一致,即有瓣类聚为一类,无瓣类聚为一类。在有瓣昆虫类群中,角蝇属的西方角蝇和截脉角蝇先聚在一起,再与其同科中的家蝇共聚在一支上;而寄蝇科的两种寄蝇、花蝇科的甘蓝地种蝇和舌蝇科的蛟舌蝇依次与西方角蝇、截脉角蝇和家蝇聚在一起。在无瓣昆虫类群中,地中海实蝇与墨西哥按实蝇聚在一起,果蝇与拟果蝇聚在一起,而后两者聚为一类。综上所述,18S rDNA基因序列中最保守同源区适用于研究不同双翅目昆虫物种间的系统发育关系。
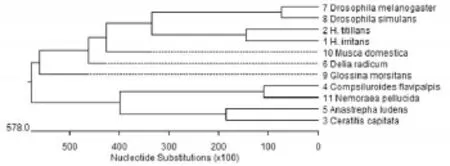
图2 11种双翅目昆虫18S rDNA全序列构建的系统发育树
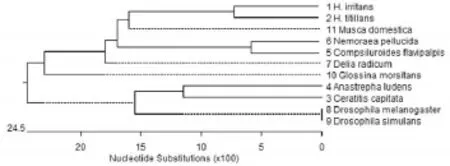
图3 11种双翅目昆虫18S rDNA最保守同源区构建的系统发育树
核酸序列中碱基组成偏异会对系统发育树的重建产生一定的影响[11],本研究中11种双翅目昆虫18S rDNA基因序列的4种碱基含量基本相似,组成上无偏异,均表现出A+T含量高于G+C含量。故本研究以18S rDNA基因序列最保守同源区为靶分子重建系统发育树未受碱基组成偏异的影响,可较为真实地在分子水平上反映双翅目各科昆虫的进化情况。
[1]Lorena T,Consuelo A,Nieves A,et al.Identification ofmicroor⁃ganisms in partially fed female horn flies,Haematobia irritans[J]. Parasitol Res,2012,111(3):1391-1395.
[2]赵治国.我国骆驼斯氏副柔线虫病传播媒介的研究[D].呼和浩特:内蒙古农业大学博士论文,2010.
[3]刘诚刚,杜智恒,白秀娟.野生乌苏里貉18S rDNA全序列的克隆测序及系统进化树分析[J].中国畜牧兽医,2012(2):117-118.
[4]魏国清,代君君,刘朝良,等.家蚕核糖体18SRNA基因的序列分析及分子系统学研究[J].经济动物学报,2006,10(3):151-155.
[5]李海超,乔奇,原国辉,等.烟夜蛾18S rDNA的克隆及序列分析[J].动物分类学报,2009,34(4):816-822.
[6]王海亭,贾月丽,程晓东,等.亚洲玉米螟核糖体18S rRNA基因的序列分析及分子系统学研究[J].河南农业大学学报,2010,44(2):85-190.
[7]Neeb J,Van de peer Y,Hendricks L,et al.Compilation of small ribosomal subunit RNA sequences[J].Nucleic Acids Res,1990,18(Suppl):2237-2317.
[8]Kjer K M.Aligned 18S and insect phylogeny[J].Syst Biol,2004,53(3):506-514.
[9]王瑛,陈晓峰,刘伟,等.棉铃虫18S核糖体RNA基因的序列分析及其分子系统学[J].昆虫学报,1999,42(3):241-247.
[10]范滋德.中国动物志昆虫纲(第四十九卷,双翅目:蝇科)[M].北京:科学出版社,2008.
[11]Lockhart pJ,Howe C J,Bryant D A,et al.Substitutional bias confounds inference of cyanelle origins from sequence data[J].J Mol Evol,1992,34(2):153-162.
The 18S rDNA cloning andmolecular systematics study of Haematobia irritans and H.titillans
SHIHong-lei,DENGQiao,YANG Lian-ru,YANG Xiao-ye,WANGRui,ZHENGWen-qing,YANG Bo
(College of Veterinary Medicine,Inner Mongolia Agricultural University;Key Laboratory of Clinical Diagnosis and Treatment Technology in Animal Disease,Ministry of Agriculture,Hohhot 010018,China)
In order to get 18S rDNA sequence and molecular evolution of diptera insects Haematobia irritans and H.titillans,we sequenced the 18S rDNA of horn flies.Then the sequence was blasted in the NCBI,and compared with the 18S rDNA of the other nine insects of Diptera in GenBank to find out their homologous regions and variable regions.Phylogenetic treewas construct⁃ed by using the complete sequence and themost conserved homologous regions.The results indicated that the 18S rDNA sequence length of Haematobia irritans and H.titillans was 1 984 bp.The homology between them was 96.4%and there were 73 recognition sites.The homology of the two horn flies with the H.irritans reported in GenBank was 99.1%and 95.7%respectively.There were three highly conserved homologous regions in the 18S rDNA gene sequences of 11 kinds of diptera insects,which was equivalent to the sequence from 320 bpto 693 bp,848 bpto 1 181 bpand 1 606 bpto 1 849 bpin 18S rDNA gene sequences of Haematobia ir⁃ritans respectively,while the first section of the homologous regionswas themost conservative.The 18S rDNA gene sequences of H.titillans were reported for the first time in the world in this article,and it proved the 18S rDNA gene sequence of H.irritans was highly conserved.So itmaybemore scientific to classify the 11 kinds of diptera insects by the phylogenetic tree,which was con⁃structed by using the most conservative homologous region,than the one constructed by the whole sequence,itwas in accordance with the resultof the traditionalmorphological classification.
Haematobia irritans;Haematobia titillans;18S rDNA;Sequence Analysis
s:YANG Lian-ru;YANG Xiao-ye
S852.74+3
A
0529-6005(2015)12-0036-03
2015-03-02
国家自然科学基金(31160502);公益性行业(农业)科研专项(201303037)
史红蕾(1988-),女,硕士生,主要从事兽医公共卫生学研究,E-mail:14747345688@163.com
杨莲茹,E-mail:lianruyang122@163.com;杨晓野,E-mail:xiaoyeyang122@sohu.com
猜你喜欢
玩具世界(2022年3期)2022-09-20
基层中医药(2022年4期)2022-07-22
汉字汉语研究(2021年2期)2021-08-30
甘肃教育(2020年2期)2020-09-11
农村青少年科学探究(2020年5期)2020-08-18
汉字汉语研究(2019年2期)2019-08-27
小学生必读(低年级版)(2018年11期)2018-03-13
新民周刊(2018年8期)2018-03-02
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
饮食科学(2017年12期)2018-01-02
