
烯烃聚合用Z-N催化剂活性中心的影响因素及研究方法
2015-03-28 08:37傅智盛范志强
合成树脂及塑料 2015年1期
徐 涛,傅智盛,范志强
(高分子合成与功能构造教育部重点实验室,浙江大学高分子科学与工程学系,浙江省杭州市 310027)
Ziegler-Natta(Z-N)催化剂具有活性高,产物颗粒形态、加工性能好,成本低,稳定性高,适合大规模连续生产的优势[1-2]。Z-N催化剂的活性中心前体是通过Cl桥结合在MgCl2晶体表面特定部位的Ti物种[3]。负载型Z-N催化剂具有活性中心多分散性的特点[4-5],聚合物的宽相对分子质量分布[5]、宽化学组成分布[6-7]以及立体结构多样性[4,8-11]都起源于Z-N催化剂活性中心的多分散性。负载型Z-N催化剂中存在3~7种活性中心,但到目前为止,各种活性中心的真实数量还无法准确测量[5,10-11]。因此,开发合适的研究方法,准确测量各种活性中心的数量是进一步深入了解Z-N催化剂催化机理的关键。本文主要综述了影响Z-N催化剂活性中心的因素及其研究方法。
1 Z-N催化剂活性中心的影响因素
1.1 内给电子体
以酯类、醚类为代表的内给电子体是在催化剂制备过程中加入的,在Z-N催化剂中的主要贡献是提高聚丙烯的立构规整度。此外,内给电子体对催化剂的载Ti量和Ti分布以及MgCl2载体的微晶结构和形态都有重要影响。
使用MgCl2作载体制备催化剂时,MgCl2的(110)和(100)晶面上Mg分别与4个Cl和5个Cl配位(见图1)。内给电子体能与TiCl4竞争,优先与位于(110)晶面上酸性较强的Mg结合,减小了TiCl4与(110)晶面上的Mg接触形成无规活性中心的可能性,影响了载体上的Ti分布,提高了聚丙烯的等规指数,同时也使催化剂的载Ti量有所降低[12]。
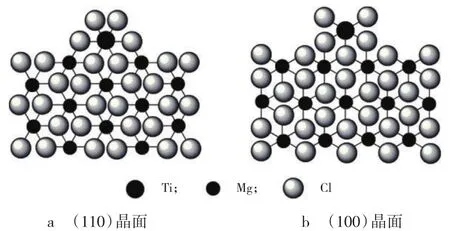
图1 TiCl4在MgCl2晶体表面可能的配位方式示意Fig.1 Plausible coordination model schematics of TiCl4 on the surface of the MgCl2 crystal
最近研究认为,内给电子体打破了(110)晶面上无规单金属活性中心和等规双金属活性中心之间的平衡,使之向后者移动。当然,内给电子体与活性中心附近的MgCl2配位,也会改变活性中心的电化学环境和立体位阻,最终导致聚合物等规指数的提高[13]。Taniike等[14]通过分子模拟计算发现TiCl4会以单核形态优先吸附在MgCl2晶体的(110)晶面,而共吸附于此晶面上的内给电子体能增强Ti活性中心的电子密度,并使活性中心由无规向等规转变。
化合物的各种异构体作为内给电子体对活性中心的影响也有明显差异,邻苯二甲酸二甲酯中的两个羰基氧原子的距离恰好能与(110)晶面上的Mg螯合,使TiCl4在(110)晶面无法形成无规活性中心,而间苯二甲酸二甲酯和对苯二甲酸二甲酯则没有此效果[15]。Correa等[16]通过离散傅里叶变换(DFT)研究了几种内给电子体与MgCl2间的相互作用,认为1,3-二醚、烷氧基硅烷的两个氧原子间的距离较短,因此只能配位在(110)晶面的Mg上;而邻苯二甲酸盐、琥珀酸盐中的氧原子间的距离较长,能采取多种配位形式配位在(100)和(110)晶面的Mg上。
1.2 外给电子体
外给电子体的毒化作用使活性中心浓度下降,但无规活性中心浓度的降幅更明显,因而等规活性中心比例相对升高,使聚丙烯等规指数提高[17]。Sacchi等[18]研究发现,烷氧基硅烷中烷基体积越大、数目越多,毒化无规活性中心的能力就越强,活化等规活性中心的效果也越好。
1.3 共聚单体效应
Chien等[19]提出了“共聚单体效应”这一概念,在烯烃聚合过程中,共聚单体的加入会改变聚合反应速率;MgCl2负载的催化剂催化乙烯聚合时,共聚单体的加入使催化剂颗粒破碎,暴露出新的活性中心,增加了活性中心的浓度。
Kissin等[20]根据动力学和端基分析研究提出,活性中心的β—H元结效应是乙烯与α-烯烃共聚合时共聚单体活化的根本原因。乙烯插入Ti—H会形成Ti—CH2—CH2—H,由于β—H元结效应,Ti的空位被位于β位的H占据,使Ti—CH2—CH2—H很稳定,后续的乙烯插入十分缓慢,导致活性中心进入休眠状态,而活性中心与α-烯烃聚合时,H难以占据Ti空位,立即形成Ti—(CH2—CH2)n—R(R为烷基),避免了休眠中心Ti—CH2—CH2—H的形成,增加了聚合活性。
1.4 氢气对活性中心的活化作用
α-烯烃聚合时,加入氢气使聚合反应速率显著提高,说明氢气对活性中心有一定的活化作用。Kissin等[20]认为丙烯聚合时,氢气对活性中心的活化作用也是活性中心的β—H元结效应。Terano等[21]利用停流技术,能在极短时间(0.1 s)内研究丙烯聚合动力学。他们认为氢气的加入既不产生新的活性中心,也不影响链增长速率常数,但是能使休眠中心重新活化。Terano等[22]制备了超低载Ti量催化剂,采用同位素交换实验研究后发现,氢气会被具有较高立体选择性的活性中心裂解成H,参与链转移反应,但超低载Ti量催化剂上的Ti都是孤立的活性中心,不具备这种裂解能力。而通过DFT计算发现,氢气对催化剂活性的提高与中心金属的亲电性呈负相关[23]。
1.5 助催化剂烷基铝参与活性中心的形成
Terano等[24-25]采用停流技术和升温淋洗分级技术,在三乙基铝(AlEt3)存在下,研究TiCl4/邻苯二甲酸二丁酯(DBP)/MgCl2催化剂在0~600 s内活性中心的生成、失活和转化发现,在60 s以内,最高等规活性中心的形成与助催化剂密不可分,AlEt3先与TiCl4发生烷基交换反应生成少量一氯二乙基铝(AlEt2Cl),AlEt2Cl和AlEt3的Al直接通过Cl桥或乙基桥与Ti相连。
2 基于活性中心浓度测定的聚合动力学研究方法
Zerbi等[26]利用拉曼光谱对比了实验和理论计算得到的MgCl2-TiCl4复合物的结构,认为在催化剂碾磨制备过程中,至少形成了3种结构的复合物。Potapov等[27]利用傅里叶变换漫反射红外光谱研究了助催化剂烷基铝对TiCl4/DBP/MgCl2催化剂中内给电子体的影响,发现助催化剂的加入能显著移除内给电子体并吸附在MgCl2上。Tregubov等[28]则采用电子自旋共振光谱方法研究了负载型MgCl2-TiCl4催化剂中不同氧化态的孤立态和团簇态的Ti。Busico等[29]采用DFT方法研究发现,TiCl4在MgCl2表面的吸附一般发生在(110)晶面,在(104)晶面上的吸附不稳定。Correa等[30]研究了MgCl2晶体转角上TiCl4的吸附,发现从转角的(104)晶面移除MgCl2单元所耗费的能量不高,而紧邻被移除MgCl2单元的(104)晶面旁会形成(110)晶面的结构,而内给电子体的加入使这个过程变得更容易。Chadwick等[31]制备了平板型的负载型MgCl2-TiCl4催化剂,通过原子力显微镜和扫描电子显微镜能清晰地观察到聚合物在MgCl2各类晶面上的生长。Terano等[32]制备了极低载Ti量的模型催化剂,发现这类催化剂上的Ti基本以孤立态存在,随着载Ti量增加逐渐形成团簇态的Ti。
目前比较常用的测定活性中心浓度的方法有动力学方法、放射性同位素标定法和化学淬灭法等。
2.1 动力学方法
动力学方法主要是将聚合物分子链的数目与聚合时间或产量作图,外推至聚合时间为0或产量为0,通过计算得到活性中心数,进而计算得到链增长速率最大值和活性中心数最大值。
采用停流技术能准确测定极短聚合时间内的活性中心数和微观动力学参数[24]。由于聚合时间极短,可以认为在此时间段内活性中心数基本不变,也不发生链转移和链终止,聚合物产率和相对分子质量随时间呈线性增长,基本能忽略副反应,得到最初期活性中心的信息;但该方法要求苛刻,由于活性中心形成时间极短,催化剂和助催化剂必须迅速混合并且分散均匀,单体要充分溶解在溶剂中,转化率要极低,溶液流动速度要恒定,聚合终止必须快速且充分,而且需要一定量的聚合产物进行分析。由于催化剂消耗量极大,实验操作复杂,不能跟踪动力学参数随聚合时间的变化,停流技术局限性较大。
Terano等[33]采用停流技术结合升温淋洗分级的方法针对MgCl2负载Z-N催化剂的总活性中心浓度、链增长速率常数和不同级分的质量分数进行研究后提出了三中心模型,解释了活性中心的立体定向能力的本质成因。
2.2 放射性同位素标定法
放射性同位素标定法就是将用同位素标记的化合物与活性中心反应,通过仪器等手段得到被同位素标记的物质数目,进而计算活性中心数。在Z-N催化剂的研究中,应用最广泛的是放射性14CO标记方法。CO能在活性中心的Ti上配位,插入增长的聚合物分子链,从而形成被14C标记的无法再插入烯烃分子的末端基团。不过CO与活性中心的反应极其复杂,涉及到可逆性和副反应,用此方法解释一些原因时要考虑多方面的因素。
Bukatov等[10]对DBP和2,2-二异丁基-1,3-二甲氧基丙烷进行了深入研究,他们利用放射性同位素标定法研究了乙烯和丙烯聚合的活性中心浓度和链增长速率常数,发现等规活性中心的链增长速率常数比无规活性中心的高,加入内给电子体降低了无规和低等规活性中心的链增长速率常数,对等规活性中心没有影响,外给电子体的加入提高了活性中心的立体定向能力,高等规活性中心和休眠中心的比例都有所提高。
2.3 化学淬灭法
化学淬灭法是采用反应性化合物淬灭聚合,再通过化学方法标定得到活性中心浓度,常用试剂有氚代甲醇、CS2和酰氯等。采用氚代甲醇淬灭聚合法研究均相和非均相体系都比较有效,但此法只能测定聚合初期的活性中心浓度,无法追踪其浓度变化。CS2淬灭聚合法的机理与14CO标记法相同,CS2能与带有增长链和空位的过渡金属配位插入。氚代甲醇和CS2都不可避免地会与部分助催化剂或者聚合物分子链发生副反应,影响结果的准确性。
上述方法的可靠性尚待商榷,无法推广应用。本课题组[34-35]首次采用乙酰氯(CH3COCl)作淬灭剂,结合紫外一阶导数法测定了TiCl3-烷基铝催化1-辛烯聚合体系中插入到活性链上的羰基数,进而得到活性中心数。为进一步解释CH3COCl与活性中心可能发生的反应,用三异丁基铝活化TiCl3得到活性中心 Ti—CH2CH(CH3)2,然后与CH3COCl反应得到4-甲基-2-戊酮。该方法能追踪聚合过程中总活性中心的动态变化,但只适用于聚合物易溶于有机溶剂的情况,而且烷基铝也会与CH3COCl和4-甲基-2-戊酮反应使结果产生误差。为避免以上问题,本课题组[36]采用2-噻吩酰氯(TPCC)作淬灭剂,其与活性中心反应后会在聚合物分子链末端接入含硫基团(反应机理见图2),只要测定聚合物中的硫含量就能通过计算转化为活性中心浓度。
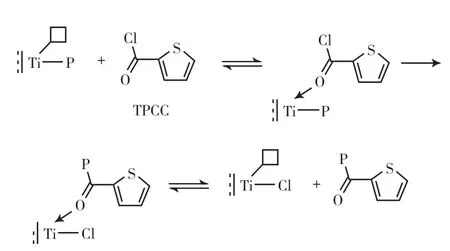
图2 TPCC与活性中心的反应机理Fig. 2 Possible quenching reaction mechanism of the active centers and TPCC
本课题组以TPCC为淬灭剂,研究了负载型MgCl2-TiCl4催化剂催化丙烯聚合初期的活性中心数,发现n(TPCC)∶n(Al)≥2时,淬灭时间小于10 min就能有效地在增长链上接入特征基团。该方法同样适用于聚合物不溶于有机溶剂的情形,与烷基铝的副反应可以忽略不计。
3 结语
Z-N催化剂相比于其他催化剂优势明显,但由于Z-N催化剂的结构和反应机理高度复杂,研究其微观机理仍面临很多瓶颈,如负载于MgCl2晶体表面的活性中心性质活泼,难以用现有表征手段直接研究;活性中心的休眠态结构、形成以及转变机理还有待进一步研究;分子模拟所用活性中心模型过于简单,无法很好解释一些实验现象。要解决上述问题,需要针对Z-N催化剂催化烯烃聚合的微观机理开展更系统、更深入的基础研究,开发更先进的表征、研究手段,从而在催化剂研制和聚合工艺领域取得突破。
[1] Qiao Jinliang,Guo Meifang,Wang Liangshi,et al. Recent advances in polyolefin technology[J]. Polym Chem,2011,2(8):1611-1623.
[2] Busico V. Metal-catalysed olefin polymerisation into the new millennium:a perspective outlook[J]. Dalton Trans,2009(41):8794-8802.
[3] Seth M,Margl P M,Ziegler T. A density functional embedded cluster study of proposed active sites in heterogeneous Ziegler-Natta catalysts[J]. Macromolecules,2002,35(20):7815-7829.
[4] Sacchi M C,Fan Zhiqiang,Forlini F,et al. Use of different alkoxysilanes as external donors in MgCl2-supported Ziegler-Natta catalysts to obtain propene/1-butene copolymers with different microstructure[J]. Makromol Chem Phys,1994,195(8):2805-2816.
[5] Fan Zhiqiang,Feng Linxian,Yang Shilin. Distribution of active centers on TiCl4/MgCl2catalyst for olefin polymerization[J]. J Polym Sci Part A:Polym Chem,1996,34(16):3329-3335.
[6] Fan Zhiqiang,Sacchi M C,Locatelli P. Effects of external donor on active center distribution of supported Ziegler-Natta catalyst[J]. Chin J Polym Sci,1997,15(3):217-225.
[7] Kissin Y V. Multicenter nature of titanium-based Ziegler-Natta catalysts:comparison of ethylene and propylene polymerization reactions[J]. J Polym Sci Part A:Polym Chem,2003,41(12):1745-1758.
[8] Kissin Y V,Chadwick J C,Mingozzi I,et al. Isoselectivity distribution of isospecific centers in supported titanium-based Ziegler-Natta catalysts[J]. Macromol Chem Phys,2006,207(5):1344-1350.
[9] Ribour D,Monteil V,Spitz R,et al. Exploring pathways to reduce the distribution of active sites in the Ziegler-Natta polymerization of propylene[J]. J Polym Sci Part A:Polym Chem,2007,45(17):3941-3948.
[10] Bukatov G D,Zakharov V A,Barabanov A A. Mechanism of olefin polymerization on supported Ziegler-Natta catalysts based on data on the number of active centers and propagation rate constants[J]. Kinetics and Catalysis,2005,46(2):166-176.
[11] Nishiyama I,Liu Boping,Matsuoka H,et al. Kinetic evaluation of various isospecific active sites on MgCl2-supported Ziegler catalysts[J]. Macromol Symp,2003,193(1):71-80.
[12] Hu Youliang,Chien J C W. Superactive and stereospecific catalysts. I. Structures and productivity[J]. J Polym Sci Part A:Polym Chem,1988,26(8):2003-2018.
[13] Taniike T,Terano M. Coadsorption and support-mediated interaction of Ti species with ethyl benzoate in MgCl2-supported heterogeneous Z-N catalysts studied by density functional calculations[J]. Macromol Rapid Commun,2007,28(18/19):1918-1922.
[14] Taniike T,Terano M. Coadsorption model for first-principle description of roles of donors in heterogeneous Ziegler-Natta propylene polymerization[J]. J Catal,2012,293:39-50.
[15] Kakkonen H J,Pursiainen J,Pakkanen T A,et al. TiCl4diester complexes-relationships between the crystal-structures and properties of Ziegler-Natta catalysts[J]. J Organomet Chem,1993,453(2):175-184.
[16] Correa A,Piemontesi F,Morini G,et al. Key elements in the structure and function relationship of the MgCl2/TiCl4/Lewis base Ziegler-Natta catalytic system[J]. Macromolecules,2007,40(25):9181-9189.
[17] Chien J C W,Weber S,Hu Youliang. Magnesium chloride supported catalysts for olefin polymerization. ⅩⅠⅩ. Titanium oxidation states,catalyst deactivation,and active site structure[J]. J Polym Sci Part A:Polym Chem,1989,27(5):1499-1514.
[18] Sacchi M C,Forlini F,Tritto I,et al. Activation effect of alkoxysilanes as external donors in MgCl2-supported Ziegler-Natta catalysts[J]. Macromolecules,1992,25(22):5914- 5918.
[19] Chien J C W,Nozaki T. Ethylene-exane copolymerization by heterogeneous and homogeneous Ziegler-Natta catalysts and the ″comonomer″ effect[J]. J Polym Sci Part A:Polym Chem,1993,31(1): 227-237.
[20] Kissin Y V,Rishina L A.Kinetics of propylene and ethylene polymerization reactions with heterogeneous Ziegler-Natta catalysts:recent results[J]. Polym Sci Ser A,2008,50(11):1101-1121.
[21] Mori H,Endo M,Tashino K,et al. Study of activity enhancement by hydrogen in propylene polymerization using stoppedflow and conventional methods[J]. J Mol Catal A:Chem,1999,145(1/2):153-158.
[22] Kouzai I,Wada T,Taniike T,et al. Hydrogen effects for propylene polymerization with ultra low TiCl3loading MgCl2-supported catalyst[J]. Macromolecular Symposia,2007,260(1):179-183.
[23] Coussens B B,Budzelaar P H M,Friederichs N. A systematic computational study of electronic effects on hydrogen sensitivity of olefin polymerization catalysts[J]. J Phys:Condens Matter,2008,20(6):064241.
[24] Liu Boping,Nitta T,Nakatani H,et al. Stereospecific nature of active sites on TiCl4/MgCl2Ziegler-Natta catalyst in the presence of an internal electron donor[J]. Macromol Chem Phys,2003,204(3):395-402.
[25] Nakatani H,Nitta T,Liu Boping,et al. Formation,deactivation and transformation of stereospecific active sites on TiCl4/dibutylphthalate/Mg(OEt)2catalyst induced by short time reaction with Al-alkyl cocatalyst[J]. J Mol Catal A:Chem,2002,180(1/2):25-34.
[26] Brambilla L,Zerbi G,Piemontesi F,et al. Structure of MgCl2-TiCl4complex in co-milled Ziegler-Natta catalyst precursors with different TiCl4content:experimental and theoretical vibrational spectra[J]. J Mol Catal A:Chem,2007,263(1/2):103-111.
[27] Potapov A G,Bukatov G D,Zakharov V A. DRIFTS study of the interaction of the internal donor in TiCl4/di-nbutylphthalate/MgCl2catalysts with AlEt3cocatalyst[J]. J Mol Catal A:Chem,2010,316(1):95-99.
[28] Tregubov A A,Zakharov V A,Mikenas T B. Supported titanium-magnesium catalysts for ethylene polymerization:a comparative study of catalysts containing isolated and clustered titanium ions in different oxidation states[J]. J Polym Sci Part A:Polym Chem,2009,47(23):6362-6372.
[29] D′Amore M,Credendino R,Busico V,et al. A periodic hybrid DFT approach(including dispersion) to MgCl2-supported Ziegler-Natta catalysts-1:TiCl4adsorption on MgCl2crystal surfaces[J]. J Catal,2012,286:103-110.
[30] Correa A,Credendino R,Pater J T M,et al.Theoretical investigation of active sites at the corners of MgCl2crystallites in supported Ziegler-Natta catalysts[J]. Macromolecules,2012,45(9):3695-3701.
[31] Andoni A,Chadwick J C,Niemantsverdriet H J W,et al. The role of electron donors on lateral surfaces of MgCl2-supported Ziegler-Natta catalysts:observation by AFM and SEM[J]. J Catal,2007,257(1):81-86.
[32] Wada T,Taniike T,Kouzai I,et al. Propylene polymerization performance of isolated and aggregated Ti species studied using a well-designed TiCl3/MgCl2Ziegler-Natta model catalyst[J].Macromol Rapid Commun,2009,30(11):887-891.
[33] Liu Boping,Nitta T,Nakatani H,et al. Precise arguments on the distribution of stereospecific active sites on MgCl2-supported Ziegler-Natta catalysts[J]. Macromol Symp,2004,213(1):7-18.
[34] 范志强,封麟先,杨士林. 用乙酰氯淬灭法测定Ziegler-Natta催化剂的活性中心数[J]. 高分子学报,1991(4):503-507.
[35] Fan Zhiqiang,Zhang Letian,Xia Shengjie,et al. Effects of ethylene as comonomer on the active center distribution of 1-hexene polymerization with MgCl2-supported Ziegler-Natta catalysts[J]. J Mol Catal A:Chem,2011,351:93-99.
[36] Shen Xianrong,Hu Jie, Fu Zhisheng,et al. Counting the number of active centers in MgCl2-supported Ziegler-Natta catalysts by quenching with 2-thiophenecarbonyl chloride and study on the initial kinetics of propylene polymerization[J].Catalysis Communication,2013,30:66-69.
猜你喜欢
火炸药学报(2022年5期)2022-11-04
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
物理实验(2019年7期)2019-08-06
当代陕西(2019年6期)2019-04-17
航空材料学报(2019年2期)2019-04-15
西南石油大学学报(自然科学版)(2018年6期)2018-12-26
物理学报(2018年22期)2018-12-18
食品工业科技(2014年15期)2014-03-11
无机化学学报(2014年4期)2014-02-28
郑州大学学报(理学版)(2013年2期)2013-03-11
