
共聚合改性可生物降解PBS
2015-03-28 08:36谢文杰周晓明
合成树脂及塑料 2015年1期
谢文杰,周 磊,周晓明*
(1.天津科技大学材料科学与化学工程学院,天津市 300457;2.天津出入境检验检疫局,天津市 300308)
聚丁二酸丁二酯(PBS)力学性能优异,具有良好的加工性,其原料既可以是石油资源,也可以通过淀粉或低聚糖发酵得到,一直备受关注[1];但PBS的结晶度高达40%~60%,致使其在自然界中生物降解速率较慢。因此,可以通过共聚合或共混方法改善PBS的性能[2]。本工作通过在PBS主链中引入具有立体结构的1,4-环己烷二甲醇(CHDM),合成了1,4-丁二醇(BD)-1,4-丁二酸(SA)-CHDM共聚物[P(BS-co-CHDM)],以期达到改善PBS力学及降解性能的目的。由于聚合物在实际加工过程中的结晶行为对其降解性能及力学性能等均有明显影响[3],因此,通过差示扫描量热法(DSC)探究PBS及P(BS-co-CHDM)的非等温结晶行为非常有必要。本研究分别采用Jeziorny法和莫志深法分析P(BS-co-CHDM)的非等温结晶动力学过程,并对其晶体结构、结晶形态及力学性能进行了研究。
1 实验部分
1.1 主要试剂
SA,BD,均为分析纯,国药集团化学试剂有限公司生产;CHDM,分析纯,阿法埃莎(天津)化学有限公司生产;SnCl2,分析纯,天津市北方天医化学试剂厂生产;对甲苯磺酸,分析纯,天津市赢达稀贵化学试剂厂生产。
1.2 主要设备
NETZSCH DSC 204F1型差示扫描量热仪,德国耐驰仪器制造有限公司生产;TZL-30F型广角X射线衍射仪,丹东通达科技有限公司生产;DRX-400MHz型核磁共振波谱仪,德国Bruker公司生产;BK-POLR型偏光显微镜,重庆奥特光学仪器有限公司生产;CMT4503型电子万能试验机,深圳市新三思材料检测有限公司生产。
1.3 P(BS-co-CHDM)的合成
在四颈烧瓶中,加入不同配比的反应物,采用低温酯化和高温缩聚合两步反应,制备了一系列P(BS-co-CHDM)。n(BD)∶n(SA)为1.05∶1.00,n(BD)∶n(CHDM)分别为90.0∶10.0,80.0∶20.0,70.0∶30.0,同时加入适量催化剂SnCl2和对甲苯磺酸,氮气气氛,在140 ℃条件下反应1~2 h,完全除去酯化反应产物——水;将温度降至100 ℃,再加入适量SnCl2,逐渐升温至220 ℃,在低于50 Pa的真空条件下反应2~3 h,直至反应物黏度达到最大值。反应完毕后,在氮气保护的条件下将反应物倒入三氯甲烷中溶解,用甲醇沉淀并反复洗涤后,在50 ℃烘箱中干燥24 h,得到白色产物P(BS-co-CHDM),其结构见图1。

图1 P(BS-co-CHDM)的结构Fig.1 Structure of P(BS-co-CHDM)
1.4 测试与表征
核磁共振氢谱(1H-NMR)测试:氘代氯仿为溶剂,四甲基硅烷为内标。DSC分析:先以20.0 ℃/min升至150 ℃,恒温3 min消除热历史后,分别以20.0,10.0,5.0,2.5 ℃/min降温到-20 ℃。力学性能按GB/T 1040.3—2008测试,拉伸速度50 mm/min。广角X射线衍射(WAXD)分析:扫描速率为8(°)/min,扫描区间为10°~50°。
2 结果与讨论
2.1 P(BS-co-CHDM)的结构表征
从图2看出:化学位移(δ)为4.1,1.7的峰分别为BD链段上的CH2(位置2)和CH2(位置3)的质子共振峰;δ为2.6处是SA链段上的CH2(位置1)的质子峰,δ为3.9~4.0是CHDM链段上的CH2(位置4)的质子峰。根据位置2和位置4处质子峰面积比可计算出P(BS-co-CHDM)中n(BD)∶n(CHDM)。投料时n(BD)∶n(CHDM)为80.0∶20.0,计算得出P(BS-co-CHDM)中n(BD)∶n(CHDM)为78.6∶21.4,接近投料时的n(BD)∶n(CHDM)。
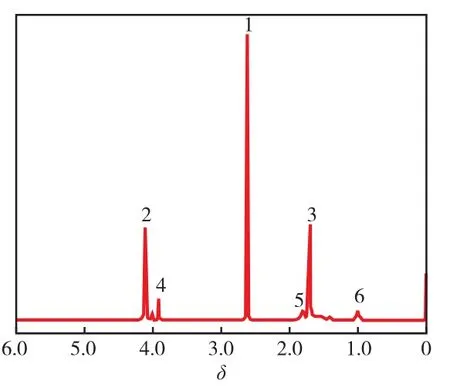
图2 P(BS-co-CHDM)的1H-NMR谱图Fig.2 1H-NMR spectrum of P(BS-co-CHDM)
2.2 P(BS-co-CHDM)的非等温结晶行为
从图3可看出:所有P(BS-co-CHDM)试样的结晶峰均随降温速率(β)升高而显著变宽,结晶峰位置向低温方向移动。从图3d看出:n(BD)∶n(CHDM)为70.0∶30.0时,P(BS-co-CHDM)结晶速率较慢,没有明显的结晶放热峰,因此,本研究未对其进行结晶动力学分析。

图3 PBS及P(BS-co-CHDM)在不同β条件下的DSC曲线Fig.3 DSC curves of PBS and P(BS-co-CHDM) at different cooling rates
从表1可以看出:在不同β条件下,n(BD)∶n(CHDM)为90.0∶10.0时,P(BS-co-CHDM)的结晶温度(θc)与PBS接近甚至超过PBS;而n(BD)∶n(CHDM)为80.0∶20.0时,P(BS-co-CHDM)的θc远低于纯PBS。这说明CHDM含量较低时对P(BS-co-CHDM)的θc影响不大。
2.3 P(BS-co-CHDM)的非等温结晶动力学
非等温条件下的相对结晶度(Xt)由式(1)计算得到。

式中:θ0,θ∞分别指结晶起始温度和终止温度,dHc/dθ表示热流变化率。在非等温条件下,某时刻(t)和温度(θ)的关系见式(2)。

由式(1)与式(2)可得Xt与t的关系(见图4)。

表1 PBS及P(BS-co-CHDM)在不同β条件下的DSC参数Tab.1 DSC parameters of PBS and P(BS-co-CHDM)at different cooling rates

图4 PBS及P(BS-co-CHDM)的Xt与t的关系Fig.4 Plots of relative crystallinity versus crystallization time for PBS and P(BS-co-CHDM)
从图4看出:PBS及P(BS-co-CHDM)在非等温结晶时,随β的降低,达到相同Xt需要更长时间。
2.3.1 Jeziorny方法分析
聚合物DSC等温结晶过程一般用Avrami方程[4]描述,见式(3)。
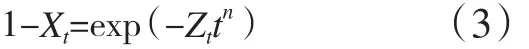
式中:Zt为结晶动力学速率常数;n为Avrami指数,与成核方式以及生长过程有关。
对式(3)两边取双对数得式(4)。
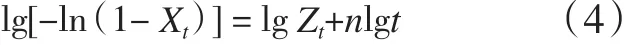
将lg[-ln(1-Xt)]对lgt作图,在结晶初期两者呈直线关系,由直线的斜率和截距可以得到n和Zt,而在结晶后期,也就是二次结晶阶段,曲线出现偏离现象。从图5可以看出:在结晶初期曲线呈现很好的线性关系。用Jeziorny拟合得到的n,lgZt见表2。
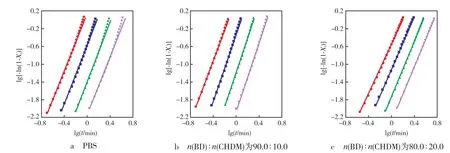
图5 PBS及P(BS-co-CHDM)结晶初期lg[-ln(1- Xt)]与lgt的关系Fig.5 Plots of lg[-ln(1-Xt)] versus lgt for PBS and P(BS-co-CHDM) in primary crystallization stage

表2 用Jeziorny方法分析得到的结晶初期的动力学参数Tab.2 Primary crystallization kinetic parameters obtained from Jeziorny′s method
考虑到非等温结晶的特点,Jeziorny[5]提出对用Avrami方程得到的Zt用β进行校正[见式(5)]。

从表2可以看出:PBS及P(BS-co-CHDM)的n为2.00~4.00,Zc随着β的增大而增大,说明随着β的增大结晶速率也相应加快。β一定时,与纯PBS比较,P(BS-co-CHDM)的Zc随着CHDM含量的增加呈先增大再减小的趋势。t1/2常被用来衡量结晶速率的快慢,t1/2随着β的增大而减小。这可能是因为P(BS-co-CHDM)主链中引入少量CHDM立体结构,分子间作用力减弱起主导作用,有效增加了n(BD)∶n(CHDM)为90.0∶10.0时P(BS-co-CHDM)的自由体积,促使分子链的空间运动能力增强;随着CHDM含量的进一步增加,PBS分子链的规整性破坏严重,此时空间位阻增大起主导作用,降低了分子链的运动能力,从而导致n(BD)∶n(CHDM)为80.0∶20.0时,P(BS-co-CHDM)的结晶速率降低。
2.3.2 莫志深方法分析
为了更有效地描述聚合物的非等温结晶动力学,莫志深[6]提出将Avrami方程与Ozawa方程相结合的处理方法,其方程见式(6)。

整理式(6)可得式(7)。

式中:a=n/m,m为Ozawa指数。动力学参数F(T)=[K(T)/Zt]1/m,其物理意义为某一温度条件下,单位结晶时间内体系达到某一结晶度所需β。F(T)越大,体系的结晶速率越低。K(T)为热力学温度(T)的函数,与成核方式、成核速率、晶核生长速率等因素相关。根据式(7),在不同Xt时的lgβ对lgt作图(见图6),拟合得到的F(T),a。从表3可看出:对于所有P(BS-co-CHDM)的非等温结晶过程,F(T)与Zc的结论一致,与测定的t1/2的变化规律相同,同时在选取的Xt范围内a变化不大。这说明用莫志深方法处理PBS及P(BS-co-CHDM)的非等温结晶动力学行为是有效的。

图6 PBS及P(BS-co-CHDM)非等温结晶时lgβ与lgt 的关系Fig.6 Plots of lgβ versus lgt for PBS and P(BS-co-CHDM) during non-isothermal crystallization

表3 莫志深方法分析得到的PBS及P(BS-co-CHDM)的结晶动力学参数Tab.3 Crystallization kinetic parameters of PBS and P(BS-co-CHDM) obtained by Mo′s method
2.4 P(BS-co-CHDM)的晶体结构及结晶形态
从图7可以看出:PBS的3个特征衍射峰对应的衍射角分别为20.1°,22.3°,23.1°,这些衍射峰分别对应(020),(021),(110)晶面。P(BS-co-CHDM)的衍射峰均向小角度偏移,但晶体结构并没有发生大的变化,均为单斜α晶型[7]。
从图8看出:随着CHDM含量的增加,P(BS-co-CHDM)的环带球晶形态消失,同时P(BS-co-CHDM)的晶体尺寸减小,说明引入具有立体结构的CHDM,影响了分子运动排列,添加量越大越难以形成规整的球晶,减小了P(BS-co-CHDM)的球晶尺寸。
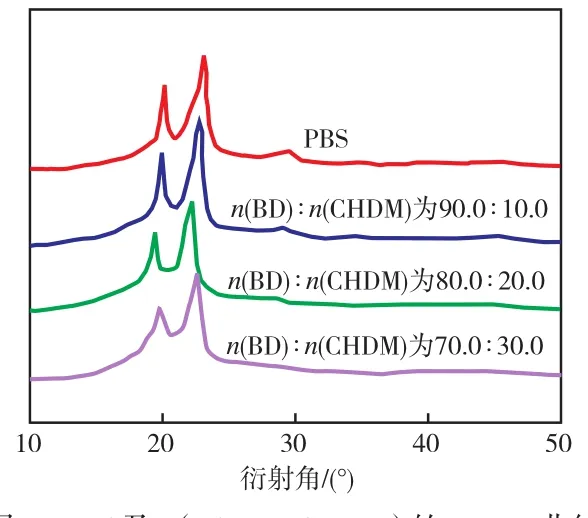
图7 PBS及P(BS-co-CHDM)的WAXD曲线Fig.7 WAXD curves of PBS and P(BS-co-CHDM)

图8 PBS及P(BS-co-CHDM)在70 ℃等温结晶2 h的偏光显微镜照片(×600)Fig.8 Polarized light microscopic photos of isothermal crystallization of PBS and P(BS-co-CHDM) at 70 ℃ for 2 hours
2.5 P(BS-co-CHDM)的力学性能
从表4可以看出:随着CHDM含量的增加,P(BS-co-CHDM)的拉伸弹性模量先增加后减小,而断裂拉伸应变显著提高。n(BD)∶n(CHDM)为90.0∶10.0时,P(BS-co-CHDM)的拉伸弹性模量达到495 MPa。n(BD)∶n(CHDM)为80.0∶20.0时,断裂拉伸应变最大,达到208.9%。这说明CHDM的加入使P(BS-co-CHDM)的柔顺性增强。

表4 PBS及P(BS-co-CHDM)的力学性能Tab.4 Mechanical properties of PBS and P(BS-co-CHDM)
3 结论
a)Jeziorny法和莫志深法均可以描述纯PBS和P(BS-co-CHDM)的非等温结晶动力学行为。与纯PBS相比,P(BS-co-CHDM)的结晶速率随CHDM含量的增加先增大再减小。
b)由于CHDM加入PBS的主链结构中,破坏了分子链的结构规整性,晶体尺寸减小,但晶体结构并没有发生太大变化。
c)CHDM的引入使P(BS-co-CHDM)的柔顺性增强,断裂拉伸应变显著提高。
[1] Ravati Sepehr,Basil D Favis. Tunable morphologies for ternary blends with poly(butylene succinate):partial and complete wetting phenomena[J]. Polymer,2013,54(13):3271-3281.
[2] 周晓明,揣成智,许磊. 聚(丁二酸丁二酯-co-衣康酸丁二酯)的合成[J]. 合成树脂及塑料,2012, 29(4):15-17.
[3] Tan Bin,Qu Jinping,Liu Liming,et al. Non-isothermal crystallization kinetics and dynamic mechanical thermal properties of poly(butylene succinate) composites reinforced with cotton stalk bast fibers[J]. Thermochimica Acta,2011,525(1/2):141-149.
[4] Avrami M. Kinetics of phase change. Ⅱ. Transformation-time relations for random distribution of nuclei[J]. J Chem Phys,1940,8(2):212-224.
[5] Jeziorny A. Parameters characterizing the kinetics of the nonisothermal crystallization of poly(ethylene terephthalate)determined by DSC[J]. Polymer,1978,19(10):1142-1144.
[6] 莫志深. 一种研究聚合物非等温结晶动力学的方法[J]. 高分子学报,2008(7):656-661.
[7] He Yisong,Zeng Jianbing,Li Shaolong,et al. Crystallization behavior of partially miscible biodegradable poly(butylene succinate)/poly(ethylene succinate) blends[J]. Thermochimica Acta,2012,529(7):80-86.
猜你喜欢
河南科技(2023年1期)2023-02-11
学与玩(2022年12期)2023-01-11
Chinese Physics B(2022年5期)2022-05-16
黑龙江交通科技(2020年5期)2020-01-13
有机氟工业(2019年2期)2019-08-12
中国塑料(2016年9期)2016-06-13
中国人兽共患病学报(2016年6期)2016-01-30
中国塑料(2015年3期)2015-11-27
中国塑料(2015年7期)2015-10-14
制造技术与机床(2015年3期)2015-01-27
