
金属原子(Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)与金富勒烯混合纳米团簇的研究
2015-03-23 11:56杨彦闯赵丽霞谢海芬罗有华
原子与分子物理学报 2015年2期
张 辰, 杨彦闯, 赵丽霞, 谢海芬, 罗有华
(华东理工大学, 上海 200237)
金属原子(Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)与金富勒烯混合纳米团簇的研究
张 辰, 杨彦闯, 赵丽霞, 谢海芬, 罗有华
(华东理工大学, 上海 200237)
运用密度泛函理论,对金属原子(Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)与金富勒烯混合纳米团簇的结构和性质进行了详细研究.在研究结果中,发现了Au31Ba纳米材料,结构比金富勒烯还要稳定,又具有较高的化学活性,可以用来做优良的催化剂.
金富勒烯; 金属原子; 密度泛函理论
1 引 言
原子分子团簇广泛存在于自然和人类实践活动中,涉及到众多过程和现象,如催化、相变、晶体生长、薄膜形成和临界现象等,构成物理和化学两大学科的交汇点.1985年,Kroto等人[1]在模拟宇宙长链碳分子的生长研究中发现了C60团簇.1990年,一个从事星际化学研究的小组[2]获得了C60晶体的结构:高对称性的中空笼状结构,称为富勒烯.该团簇不仅结构稳定,化学稳定性也很高.由于C60的出现,笼形团簇材料以奇异的特性和广阔的应用前景受到人们的极大关注,成为全世界纳米材料研究的热点.2004年,复旦大学龚新高教授[3]和芬兰的Johansson等人[4]从理论上预言了第一个金属纳米团簇的笼子结构—金富勒烯.该团簇由32个金原子组成,以三角形为单元形成了接近完美的菱形三十面体结构,具有Ih对称性和球型芳香性.
与团簇相键合的复合团簇通常被称为团簇的衍生物.在以往的研究中对C60衍生物的研究很多,而对Au32衍生物的研究非常少.本文在Au32的高对称性的中空笼状结构的基础上,掺杂碱金属原子,研究它们之间键合的特性,以及形成的新团簇的性质,期望为制备纳米新材料提供重要的参考.
2 计算方法
在本文中,计算全部采用基于密度泛函理论的DMol3程序包[5-6]来完成.考虑到金原子的相对论效应,采用了以傅立叶变换为基础的相对半芯赝势(DSPP)来处理金原子间的芯问题,电子的交换关联作用取广义梯度近似(GGA)处理, 价电子采用DND基组,自旋非限制计算,几何优化时的总能量和电子密度的收敛标准为10-6Hartree,原子的最大位移是0.005 Å.为了保证得到的是最低能量结构,我们对研究的团簇进行了频率优化,只要没有虚频,就能保证结果的准确性.
为了验证方法的可靠性,我们计算了金二聚体(Au2)的平均结合能、频率和键长,并与实验结果进行对比.计算结果表明,Au2的平均结合能 1.11eV, 频率180cm-1和键长2.544 Å,与实验值(1.15 eV ,191 cm-1,和 2.47 Å)[7]符合的很好.另外,在相同的条件下,计算得到的Au7、Au19、和 Au20的最低能量结构与实验上得到的结构一致[8](如图1所示).因此,在本文中,用GGA/PBE的方法来计算金团簇的结构和性质是可靠的.

图1 Au7、Au19、和 Au20 纳米团簇的最低能量结构和它们的对称性
3 结果与讨论

图2 Au32纳米团簇的中空笼状结构和它的对称性
Fig.2 Hollow cage-like structure of Au32nanoclusters and its symmetry
采用上面的方法,我们得到了Au32的Ih对称的中空笼状结构.在Au32结构的基础上,通过替换,我们得到了Au31M的稳定结构.通过分析发现,碱金属原子没有改变原先团簇的笼状结构,但降低了对称性.表1中给出了结构中Au-M的键长,与Au32中的Au-Au的键长(2.779 Å)比较,发现除了锂原子,Au-M中的平均键长要长一些,说明碱金属原子与金原子的相互作用力要比金原子与金原子的弱一些.从平均结合能的数据上分析, 掺杂团簇中原子间的结合能要比Au32中原子间的结合能略低一些,也说明Au-M间的相互作用力比Au-Au间的相互作用力弱.
为了进一步分析团簇的稳定性,计算了Au31M和Au32团簇的平均结合能,方程定义如下:
Eb(Au31M)=[31E(Au)+E(M)-E(Au31M)]/(31+1)
(1)
Eb(Au32)=[32E(Au)-E(Au32)]/(31+1)
(2)
这里,E(Au)、E(M)和E(Au32)分别代表了最稳定的Au、M和Au32团簇的总能量.
在图4中,我们画出了平均结合能的示意图,并在表1标出了Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba, Ra)和 Au32的值.从图中可以看出,这些团簇的结合能有明显的奇偶变化,其中,Au31Li、Au31K、Au31Be、Au31Ca、Au31Ba、Au32比相邻团簇的结合能要高,而Au31Na、Au31Rb、Au31Mg、Au31Sr和Au31Ra比相邻的结合能小.特别值得注意的是,Au31Ba的平均结合能(2.472 eV)比Au32的值(2.468 eV)还要高,这说明在结构上,前者比后者更稳定.

图3 Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)纳米团簇的中空笼状结构和它们的对称性Fig.3 Hollow cage-like structures of Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)nanoclusters and their symmetries
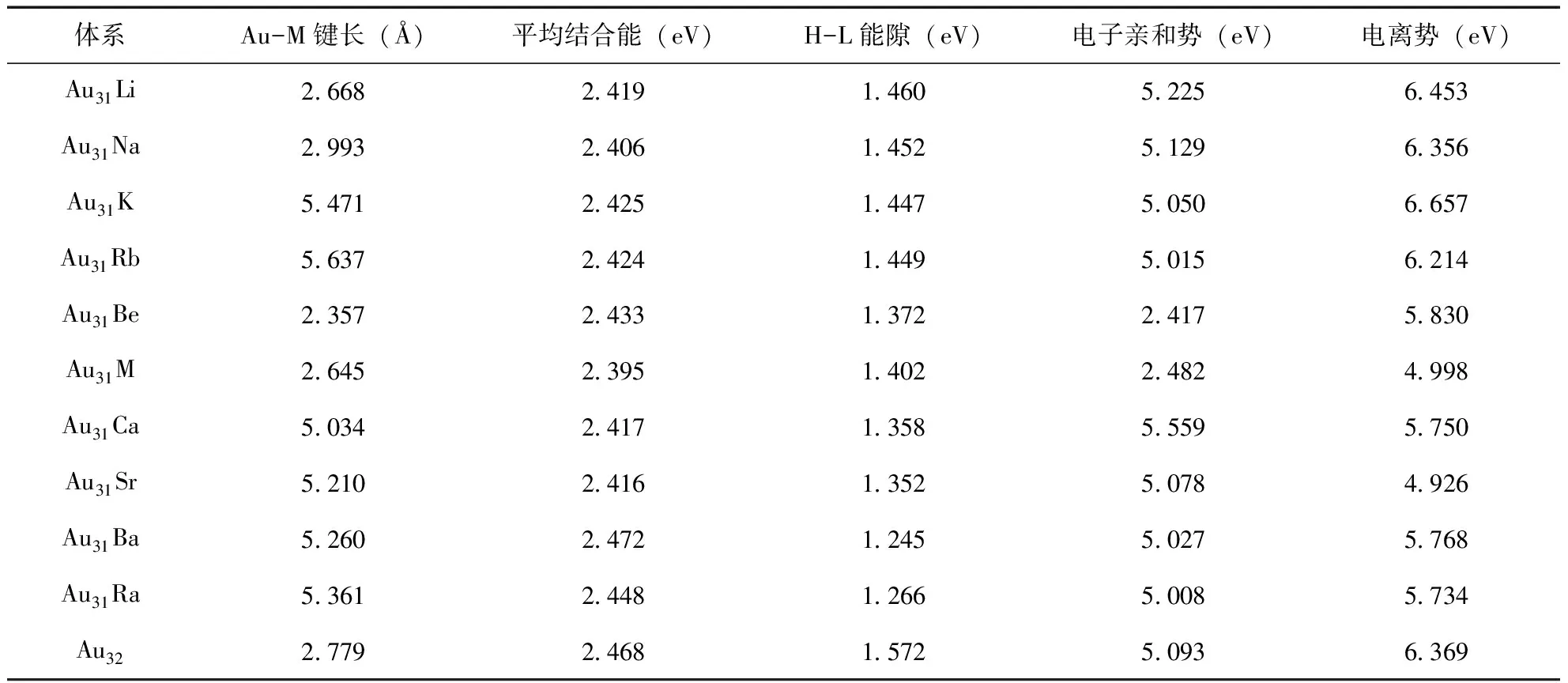
表1 Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba, Ra)纳米团簇的Au-M平均键长、平均结合能、H-L能隙、电子亲和势和电离势

图4 Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba, Ra)纳米团簇的平均结合能Fig.4 Average binding energies of Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)nanoclusters
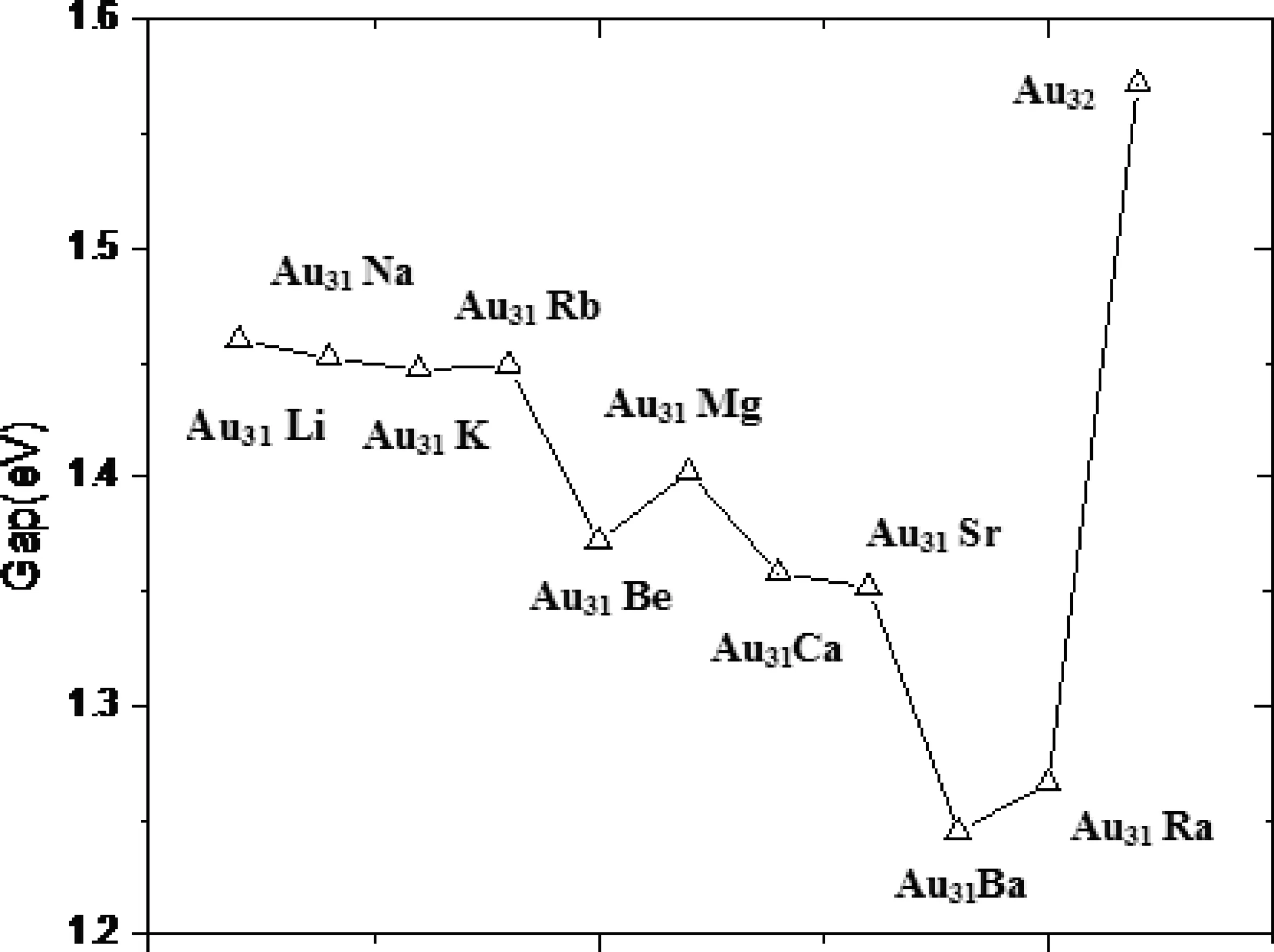
图5 Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba, Ra)纳米团簇的能隙Fig.5 Gap of Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)nanoclusters
最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的能隙反映了电子从占据轨道向未占据轨道发生跃迁的能力,在一定程度上代表分子参与化学反应活性的强弱,能隙大则说明化学活性弱,化学稳定性较强;反之则说明化学活性强,化学稳定性较弱.图5给出了Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba, Ra)团簇的笼状结构的HOMO- LUMO能隙.从图中可以看出,团簇的能隙间没有明显的变化规律,Au31Li的值最大(1.460eV), 但是没有Au32(1.572 eV)的能隙大,说明后者的化学稳定性更高.Au31Ba的能隙值最低,对应着最低的稳定性,说明其化学活性最高,前面的研究结果表明其结构稳定性是最高的.因此,在本文研究的这些纳米材料中,Au31Ba纳米团簇是最优良的催化剂材料,这将为进一步的实验研究提供重要的参考依据.
电子亲和势是了解团簇稳定性的一个重要参数,它可以用来评估团簇接受电子的能力.电子亲和势的大小在一定程度上反映了体系阴离子的稳定性,以及中性体系束缚电子的能力.图6给出了Au31M团簇的电子亲和势.从图上可以看出,体系的电子亲和势没有明显的变化趋势.其中,Au31Ca的电子亲和势最大(5.559eV), Au31Be的值最小(2.417 eV),说明前者更容易得失电子.
垂直电离能(VIP)是了解团簇化学稳定性的重要参数,代表从HOMO能级上失去一个电子的难易程度.VIP越大,HOMO能级越深,这表示团簇的化学稳定性越高.图7给出了Au31M团簇的电离能.Au31K团簇具有最大的电离势(6.657eV),Au31Sr具有最小的电离势(4.926eV), 这与他们的稳定结构密切相关.

图6 Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba, Ra)纳米团簇的电子亲和势

图7 Au31M(M= Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba, Ra)纳米团簇的电离势
4 总 结
本文利用密度泛函研究了Au31M团簇的的中空笼状稳定结构,在结构基础上计算了平均结合能、电离能和电子亲和势、最高占据分子轨道与最低未占据分子轨道之间的能隙,以便获得团簇的相对稳定性和电子性质.结果发现,Au31Ba团簇是结构最稳定的,甚至比Au32还要稳定,其化学活性又很高,是优良的催化剂.本文的研究对于正确理解纳米金材料的微观结构演变过程及制备优质的纳米金材料具有十分重要的意义.
[1] Kroto H W, Heath J R, O’Brien S C,etal.C60buckminsterfullerene [J].Nature, 1985, 318: 162.
[2] Krätschmer W, Lamb L D, Fostiropoulos K,etal. Solid C60: a new form of carbon [J].Nature(London), 1990, 347: 354.
[3] Gu X, Ji M, Wei S H,etal. AuNclusters (N=32,33,34,35): Cagelike structures of pure metal atoms [J].Phys.Rev. B, 2004, 70: 205401.
[4] Johansson M P, Sundholm D, Vaara J. Au32: a 24-carat golden fullerene [J].Angew.Chem.Int.Ed., 2004, 43: 2678.
[5] Delley B.From molecules to solids with the DMol3approach[J].J.Chem.Phys., 2000, 113(18): 7756.
[6] Hohenberg P, Kohn W.Inhomogeneous electron gas[J].Phys.Rev., 1964, 136: B864.
[7] Huber K P, Herzberg G.MolecularspectraandmolecularstructureIV.Constantsofdiatomicmolecules[M]. New York: van Nostrand Reinhold, 1979.
[8] Gruene P, Rayner D M, Redlich B,etal. Structures of neutral Au7, Au19, and Au20clusters in the gas phase [J].Science, 2008, 321: 674.
Study of metal atoms(Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)and gold fullerene mixed nanoclusters
ZHANG Chen, YANG Yan-Chuang, ZHAO Li-Xia, XIE Hai-Fen, LUO You-Hua
(East China University of Science and Technology, Shanghai 200237, China)
Metal atoms(Li,Na,K,Rb,Be,Mg,Ca,Sr,Ba)and gold fullerene mixed nanoclusters are studied by density functional theory. In the results, Au31Ba nanomaterial is found to be one excellent catalyst for high stable structure and chemical activity.Its configuration is more stable than one of gold fullerene.
Gold fullerene; Metal atoms; Density functional theory
103969/j.issn.1000-0364.2015.02.012
2014-01-06
上海市大学生创新项目(S12086)
张辰(1992—), 女, 上海市人, 本科生, 主要从事纳米材料的结构和性能研究.E-mail: chen_104@live.cn
赵丽霞. E-mail: zlx@ecust.edu.cn
O641
A
1000-0364(2015)02-0247-06
猜你喜欢
火炸药学报(2022年6期)2023-01-16
大学物理(2022年9期)2022-09-28
青岛科技大学学报(自然科学版)(2022年1期)2022-01-20
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
物理通报(2020年7期)2020-07-01
中国化妆品(2019年4期)2019-11-20
物理(2009年3期)2009-05-21
